Org. Synth. 2005, 82, 99
DOI: 10.15227/orgsyn.082.0099
1,4-DIOXENE
[2,3-Dihydro-1,4-dioxin]
Submitted by Matthew M. Kreilein, James C. Eppich, and Leo A. Paquette
1.
Checked by Christopher P. Davie and Rick L. Danheiser.
1. Procedure
A. 2-Acetoxy-1,4-dioxane. An oven-dried, 500-mL, three-necked, round-bottomed flask is equipped with a magnetic stirbar, a reflux condenser fitted with an argon inlet, a thermometer, and a 20-cm length of black rubber tubing (1/8 in thick, 1 in diameter) attached to a dry 125-mL Erlenmeyer flask wrapped in aluminum foil and containing 91.9 g (207 mmol) of lead tetraacetate (Note 1). The flask is charged with 200 mL (2.35 mol) of dioxane (Note 2) and heated with stirring at 80 °C with a “Power Light” 500 W lamp (Note 3). The lead tetraacetate is added portionwise over approximately 30 min while the temperature is increased to the reflux point. A cloudy white or tan solution develops. The reaction mixture is heated at reflux for 1 h (Note 4) and then allowed to cool to rt. Saturated NaHCO3 solution (200 mL) is carefully introduced, and the resulting mixture is filtered through a pad of Celite. The filtrate is extracted with three 100-mL portions of CH2Cl2, and the combined organic fractions are washed with 100 mL of saturated NaHCO3 solution, dried over Na2SO4, filtered, and concentrated by rotary evaporation (20 mmHg) at room temperature. The resulting oil (27.3 g) is fractionally distilled at reduced pressure (Note 5) to remove dioxane and deliver 22.0 g (73%) of 2-acetoxy-1,4-dioxane as a colorless liquid, bp 54-55 °C (0.4 mmHg) (Note 6).
B. 1,4-Dioxene. A pyrolysis apparatus is assembled as shown in the photographs below. A 50-mL, two-necked, round-bottomed flask containing 18.0-18.1 g (0.123-0.124 mol) of 2-acetoxy-1,4-dioxane is attached to a quartz tube (Note 7) packed with coarse quartz chips (ca. 1/4") and heated at 425 °C in a horizontal furnace (Notes 8, 9). The tube is attached to a U-tube that is charged with ca. 11 g of NaOH pellets. A Dewar (cold finger) condenser with a 105° angled side joint is attached to the other end of the U-tube and filled with dry ice/acetone (−78 °C). A vacuum trap, cooled in liquid nitrogen, is positioned between the cold finger and the vacuum source. A gentle flow of N2 (constricted through a needle valve) is initiated at the side neck of the flask and a gentle vacuum (ca. 265 mmHg, controlled by a digital vacuum regulator) is applied via the angled side joint of the Dewar condenser (Note 10).



The 2-acetoxy-1,4-dioxane is introduced into the vapor phase by appropriate application of a modest amount of heat from a commercial heat gun (Note 11). After the introduction of the acetate is completed (25 min), the Dewar condenser is mounted above a 25-mL, round-bottomed flask and a mixture of 1,4-dioxene and acetic acid is collected therein following warming of the cold finger bath. The liquids contained in the U-tube (Note 12) and liquid nitrogen trap are combined with the material collected from the cold finger to afford 9.4-9.8 g of a colorless to pale yellow liquid (Note 13). Sodium hydroxide pellets (4.0 g) are added, and the resulting gel-like mixture is allowed to stand for 1 h and then distilled at atmospheric pressure (Note 14). The distillate of water and 1,4-dioxene is placed in a −18 °C freezer overnight and 1,4-dioxene is obtained by decantation from the ice crystals to furnish 4.7-6.5 g (44-61%) of 1,4-dioxene as a colorless liquid (Notes 15, 16).
2. Notes
1.
Lead tetraacetate was obtained from Acros Organics (95% purity, stabilized with ca. 4%
acetic acid) and used as received. If lightly colored, the reagent was used directly. If dark-brown in color, the
lead tetraacetate was recrystallized from
acetic acid according to the procedure described in Perrin, D. D.; Armarego, W. L. F.; Perrin, D. R.
Purification of Laboratory Chemicals, 2nd ed.; Pergamon Press: Oxford, 1980; pp 497-498.
2.
Dioxane (anhydrous, 99.8%) was purchased from Alfa Aesar and distilled from
calcium hydride under argon before use.
3.
The lamp was an inexpensive model purchased from a discount distributor (
Sam's Club). The manufacturer was The Designer's Edge, 11730 N.E. 12th St., Bellevue, WA 98006.
4.
In the event that gas evolution (as evidenced by briefly turning off the Ar source and watching the bubbler) persists after the reaction mixture is heated at reflux for 1 h, refluxing is continued until gas evolution ceases.
5.
A short path distillation head attached to a receiver cow was employed with the receiving flask cooled in a dry ice/
acetone bath.
6.
In different runs, the yield ranged from
68-73%. The product exhibits the following spectroscopic properties: IR (film): 2976, 2860, 1748, 1454, 1374, 1225 cm
−1;
1H NMR
pdf (500 MHz, CDCl
3) δ 2.16 (s, 3H), 3.64 (app dt,
J = 11.8, 2.7 Hz, 1H), 3.71-3.82 (m, 4H), 4.09-4.17 (m, 1H), 5.85 (t,
J = 2.1 Hz, 1H);
13C NMR (125 MHz, CDCl
3) δ 21.3, 61.9, 66.3, 67.9, 89.4, 170.0; HRMS (ESI) Calcd for C
6H
10O
4 [M + Na]
+: 169.0471; Found: 169.0478.
7.
The dimensions of the quartz tube used by the checkers were as follows: overall length, 590 mm; length of tube between insulation inside oven, 308 mm; outside diameter, 19 mm. The tube used by the submitters had the following dimensions: overall length, 490 mm; length of tube inside oven between insulation, 280 mm; inside diameter, 13 mm.
8.
Any type of furnace may be employed, although “hot spots” should be avoided if possible. Should the bore of the oven aperture be significantly larger than the diameter of the quartz tube, the ends of the tube may be wrapped with glass wool tape to fit. The checkers used an oven manufactured by Lindberg (a unit of General Signal), Watertown, WI 53094. The submitters used an oven manufactured by Hevi Duty Electric Company, Milwaukee, WI. The checkers monitored the oven temperature using an Omegaette® Model HH308 digital thermometer equipped with a thermocouple probe that was inserted into the center of the oven. The submitters monitored the oven temperature using a Keithley Model 871 digital thermometer equipped with a ceramic temperature probe that was inserted into the center of the oven.
9.
The quartz tube was preheated at 425 °C for ≥1 h before the
2-acetoxy-1,4-dioxane was introduced.
10.
The pyrolysis setup used by the submitters did not include a liquid nitrogen trap. Their U-tube contained ca. 5 g of NaOH, and they controlled the flow of nitrogen with a fine capillary.
11.
The checkers used a heat gun with a 260-399 °C range (14 amp) manufactured by the Master Appliance Corporation, Racine, WI 53403. The submitters used a heat gun made by the same manufacturer with a 149-260 °C range (12 amp).
12.
In one run, the checkers found that gently heating the U-tube with a heat gun (after the apparatus had been disassembled) allowed more liquid to be decanted from the U-tube.
13.
1H NMR analysis indicated that this material is a ca. 75:25 mixture of
1,4-dioxene and AcOH.
14.
A short path distillation head attached to a 25-mL, round-bottomed receiver flask were used. The mixture of water and
1,4-dioxene distilled at 58-94 °C (760 mmHg).
15.
The submitters report that they obtained
10-20% pure dioxene from the Dewar condenser cold finger and additional product by distillation of the liquid decanted from the U-tube; total yield:
65%.
16.
The product exhibits the following spectroscopic properties: IR (film): 3099, 2982, 2933, 2879, 2023, 1654, 1458, 1395, 1268, 1128, 1067, 954, 898, 739 cm
−1;
1H NMR
pdf (500 MHz, CDCl
3) δ 4.07 (s, 4H), 5.96 (s, 2H);
13C NMR (125 MHz, CDCl
3) δ 64.9, 127.2; Anal. Calcd for C
4H
6O
2: C, 55.81; H, 7.02; N; Found: C, 55.69; H, 7.01.
Handling and Disposal of Hazardous Chemicals
The procedures in this article are intended for use only by persons with prior training in experimental organic chemistry. All hazardous materials should be handled using the standard procedures for work with chemicals described in references such as "Prudent Practices in the Laboratory" (The National Academies Press, Washington, D.C., 2011 www.nap.edu). All chemical waste should be disposed of in accordance with local regulations. For general guidelines for the management of chemical waste, see Chapter 8 of Prudent Practices.
These procedures must be conducted at one's own risk. Organic Syntheses, Inc., its Editors, and its Board of Directors do not warrant or guarantee the safety of individuals using these procedures and hereby disclaim any liability for any injuries or damages claimed to have resulted from or related in any way to the procedures herein.
3. Discussion
The
1,4-dioxene molecule (
1) has attracted attention for a number of years since its initial preparation by Summerbell and Bauer in 1935.
2 The symmetrical nature of its double bond and the cis orientation of the two oxygen atoms are features not commonly resident in other structural contexts. One consequence is the lowering of the first ionization potential of
1 to 8.43 eV relative to the corresponding value in dihydropyran (8.84 eV).
3 As a result, this heterocyclic building block undergoes successful [2 + 2] photocycloaddition to conjugated enones,
4 1,2-diketones,
5 and
benzene.
6 The facility with which
1 enters into the Paterno-Büchi reaction has also been documented.
7 Comparable interest has surrounded the involvement of
1,4-dioxene in thermal inverse electron demand Diels-Alder processes,
8,9 trapping with molybdenum and chromium carbene complexes,
10 [2 + 2] ketene cycloadditions,
11 and cyclopropanations with diazo compounds.
12The enol ether constitution of
1 has caused it to be regarded as a protecting group for alcohols.
13 Beyond this, conversion to
2-dioxenyllithium (
2) can be efficiently accomplished by exposure to
tert-butyllithium in THF at low temperature.
14 This organometallic intermediate has been broadly exploited by Fétizon and Hanna in synthesis,
15 and the derived stannane
3 can be smoothly acylated with acyl chlorides under conditions of
palladium catalysis.
16 Added scope is provided by the higher order cuprate
4 whose reactivity is well suited to electrophilic capture.
17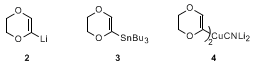
Despite the considerable promise of
1,4-dioxene in organic synthesis, only three preparative routes to
1 have been reported. More astonishing yet was the unsuitability of all three routes for the laboratory-scale acquisition of reasonable amounts of
pure reagent (1). The original pathway proceeds via the photochlorination of
p-dioxane to the 2,3-dichloro derivative
5 (
70%) followed by reductive dehalogenation with
magnesium and
iodine (
49%).
2 Entry has also been made from
diethylene glycol (
6), heating of which with a
copper chromite catalyst and KHSO
4 in the liquid phase proceeds with oxidation and cyclodehydration (
67%).
18 The complication here is the co-production of
2-p-dioxanone, a side reaction highly dependent on the proportion of KHSO
4 present. The third strategy involves the photoaddition of
phenanthrenequinone to
p-dioxane and thermal activation of the resulting
7 at 230-250 °C to liberate
1.
19The two-step process described here results in the clean formation of
1,4-dioxene free of contaminants and is therefore expected to find serviceable application in the synthesis of this useful heterocyclic intermediate. In this more practical and convenient route, advantage is taken of the rarely exploited capability
20,21 of
lead tetraacetate to engage in the acetoxylation of C-H bonds positioned at benzylic
22 and allylic sites,
23 as
well as adjacent to ethereal oxygen centers.
24 In the specific case of
p-dioxane, the eight available C-H bonds are equivalent by virtue of symmetry, thus simplifying matters considerably. The result of irradiating a refluxing solution of
lead tetraacetate in
dioxane with an inexpensive commercial 500 W light source for ca. 1 h and subsequent fractional distillation is to provide the 2-acetoxy derivative in
68-73% yield. The boiling point of this colorless liquid is sufficiently higher than that of the starting material to permit its isolation in a pure state. The flash vacuum pyrolysis step results in the smooth thermal extrusion of
acetic acid to deliver
1. When this experiment is conducted in that manner where no acetate remains unreacted, the yield of volatile
1,4-dioxene is
44-61%.
Appendix
Chemical Abstracts Nomenclature (Collective Index Number);
(Registry Number)
Lead tetraacetate:
Acetic acid, lead(4+) salt; (546-67-8)
Dioxane:
1,4-Dioxane; (123-91-1)
2-Acetoxy-1,4-dioxane:
1,4-Dioxan-2-ol, acetate; (1743-23-3)
1,4-Dioxene:
2,3-Dihydro-1,4-dioxin; (543-75-9)
Copyright © 1921-, Organic Syntheses, Inc. All Rights Reserved