Org. Synth. 2007, 84, 32
DOI: 10.15227/orgsyn.084.0032
A XANTHATE-TRANSFER APPROACH TO α-TRIFLUOROMETHYLAMINES
[2-(N-ACETYLAMINO)-4,4-DIMETHOXY-1,1,1-TRIFLUOROBUTANE]
Submitted by Fabien Gagosz and Samir Z. Zard
1.
Checked by Daniel Laurich and Alois Fürstner.
1. Procedure
A. N-(2,2,2-Trifluoro-1-hydroxyethyl)-acetamide (1). A 250-mL, two-necked, round-bottomed flask, equipped with a magnetic stir bar and fitted with a glass stopper and a condenser, is charged with 2,2,2-trifluoro-1-methoxyethanol (14.75 g, 102 mmol) (Note 1), acetamide (6.0 g, 102 mmol) (Note 1) and 1,4-dioxane (100 mL) (Note 2). The resulting solution is heated to reflux and stirred in an oil bath under argon for 2 h. The reaction mixture is then cooled to ambient temperature and transferred to a single-necked flask. The solvent is evaporated under reduced pressure and the residue dried under vacuum (25 °C, 1 mmHg) for 1 h to give 16.8 g of a colorless solid residue. This product is dissolved in tert-butyl methyl ether (20 mL) in a one-necked, round-bottomed flask. Silica gel (10 g) is added to the solution and the solvent is evaporated under reduced pressure (Note 3). The resulting adsorbate is added on top of a silica gel column (approximately 180 g), which is eluted with hexanes/ethyl acetate (3:2). The fractions containing the product are combined and evaporated to give N-(2,2,2-trifluoro-1-hydroxyethyl)acetamide (1) as a colorless solid (8.98 g, 56%) (Notes 4, 5).
B. N-1-(Chloro-2,2,2-trifluoro-ethyl)acetamide (2). A 250-mL, two-necked, round-bottomed flask is equipped with a magnetic stir bar and fitted with a glass stopper and a condenser. The top of the condenser is fitted with a T-joint allowing argon to sweep the effluent gases (HCl and sulfur dioxide) from the reaction into the exit duct of a well ventilated fume hood. The flask is charged with compound 1 (8.98 g, 57 mmol), thionyl chloride (7.1 g, 60 mmol) (Note 6) and heptane (65 mL) (Note 2). The resulting suspension is stirred and heated to 85 °C in an oil bath under argon until a clear solution has formed. Stirring is continued for 15 min before the mixture is allowed to cool to ambient temperature, whereupon the product starts to crystallize. After standing for 30 min, the crystals are collected by filtration, rinsed with heptane (2 × 25 mL) and dried under vacuum (25 °C, 1 mmHg) for 1 h to provide product 2 (9.3 g, 93%) as colorless crystals, which were used without further purification in the next step (Note 7).
C. S-(1-Acetylamino-2,2,2-trifluoroethyl) O-ethyl dithiocarbonate (3). A 500-mL, one-necked, round-bottomed flask, equipped with a magnetic stir bar, is charged with N-(1-chloro-2,2,2-trifluoro-ethyl)acetamide 2 (9.2 g, 52 mmol) and EtOH (100 mL) (Note 2). Potassium O-ethyl xanthate (9.2 g, 57 mmol) (Note 8) is added in portions over a period of 5 min. The resulting mixture is stirred at ambient temperature for 15 min (Note 9) before the reaction is quenched with water (100 mL). The solution is extracted with ether/hexanes solution (7:3, 3 × 150 mL), the combined organic layers are dried over anhydrous magnesium sulfate, filtered and evaporated under reduced pressure to provide a solid residue which is dried under vacuum (25 °C, 1 mmHg) for 1 h to give xanthate 3 (12.5 g, 91%) as a white solid (Note 10).
D. 3-Acetylamino-1-ethoxythiocarbonylsulfanyl-4,4,4-trifluorobutyl acetate (4). A flame-dried, 100-mL, two-necked, round-bottomed flask is fitted with magnetic stir bar, a glass stopper and a condenser connected to the argon line. The flask is charged with xanthate 3 (12.4 g, 47.5 mmol), vinyl acetate (5.1 mL, 54.6 mmol) (Note 11) and 1,2-dichloroethane (50 mL) (Note 11) and the resulting solution is heated under reflux in an oil bath for 15 min under argon. Four equal portions of lauroyl peroxide (473 mg each, 1.9 g overall, 10 mol%) (Notes 11–13) are added in intervals of 1.5 h to the refluxing solution. After 7 h under reflux, the solution is allowed to reach ambient temperature before the solvent is evaporated under reduced pressure to give adduct 4 as a pale yellow oil (17.8 g), which was used in the next step without further purification (Note 14).
E. N-(3,3-Dimethoxy-1-trifluoromethyl-propyl)-acetamide (5). A flame-dried, 250-mL, two-necked, round-bottomed flask is fitted with a large magnetic stir bar, a glass stopper and a reflux condenser connected to the argon line. The flask is charged with the crude radical adduct 4 (17.8 g), MeOH (100 mL) and (±)-10-camphorsulfonic acid (12.5 mol%, 1.37 g, 5.9 mmol) (Note 15). The mixture is refluxed in an oil bath for 24 h before it is cooled to ambient temperature. The solvent is evaporated under reduced pressure. The residue is dissolved in ethyl acetate (80 mL) and the organic phase is washed successively with saturated aqueous sodium bicarbonate (20 mL) and saturated aqueous sodium chloride (20 mL). The organic solvent is dried over MgSO4, filtered, and evaporated under reduced pressure. The resulting brown syrup (11.8 g) is purified by flash chromatography on silica gel (Notes 16, 17) using hexanes/ethyl acetate (2:3) as the eluent to give N-(3,3-dimethoxy-1-trifluoromethyl-propyl)-acetamide (5) as colorless crystals (4.43 g, 41%) (Note 18).
2. Notes
1.
2,2,2-Trifluoro-1-methoxyethanol was obtained from Avocado (trifluoroacetaldehyde methyl hemiacetal), tech. 90% and acetamide was obtained from Acros, 99%. The checkers used 2,2,2-trifluoro-1-methoxyethanol purchased from ABCR and acetamide purchased from Riedel de Haën.
2.
1,4-Dioxane was obtained from SDS Carlo Erba and used as received. All the solvents (petroleum ether, ethyl acetate, heptane, acetone, ethanol, methanol,
tert-butyl methyl ether) were obtained from SDS Carlo Erba and used as received. The checkers used reagent-grade solvents purchased from Acros.
3.
(E. Merck, Darmstadt, 230–240 mesh) was used. The progress of the reaction was monitored by TLC on silica gel using hexanes/ethyl acetate (3:2) as eluent. The product has an R
f = 0.15 (stained with potassium permanganate solution [
300 mL of water,
3 g of KMnO4,
20 g of K2CO3,
0.25 mL of acetic acid]).
4.
The submitters reported purification of the crude product by recrystallization:
Dichloromethane (60 mL) is added to the crude product and the resulting mixture is vigorously stirred for 10 min. The resulting white precipitate is filtered, washed twice with
30 mL of dichloromethane and dried under vacuum (25 °C, 1 mmHg) for 1 h to give a crop of pure product. The corresponding filtrate is concentrated under reduced pressure, diluted in
80 mL of ethyl acetate and washed twice with
20 mL of a half-saturated aqueous sodium chloride solution. The organic solvent is then dried over anhydrous magnesium sulfate, filtered and evaporated to give a semi-solid residue.
Dichloromethane (30 mL) is added and the mixture stirred vigorously for 10 min. The resulting white precipitate is filtered, washed twice with
10 mL of dichloromethane and dried under vacuum (25 °C, 1 mmHg) for 1 h to provide a second crop of pure product. The two crops are combined to give amide
1 (53% yield). The checkers obtained 41% of product using this purification method.
5.
The product exhibited the following properties: mp 117–119 °C;
1H NMR
pdf (MeOD, 400 MHz) δ: 2.01 (s, 3 H), 4.83 (s, 2 H), 5.71 (m, 1 H).
6.
Thionyl chloride (99.5%) was obtained from Acros and used as received; the checkers used thionyl chloride (99%) purchased from Fluka.
7.
The product exhibited the following properties: mp 85–87 °C (heptane).
1H NMR
pdf (CDCl
3, 400 MHz) δ: 2.15 (s, 3 H), 6.33 (qd,
J = 5.3, 11.0 Hz, 1 H), 7.31 (br. m, 1 H);
13C NMR
pdf (CDCl
3, 100 MHz) δ: 23.0, 60.7 (q,
J = 38 Hz), 121.8 (q,
J = 277 Hz), 170.4; IR (film): 3279, 3037, 1684, 1530, 1375, 1349, 1244, 1192, 1150, 1133, 857, 788, 690 cm
−1; HRMS (EI): Calcd for C
4H
5NOF
3Cl: 175.0011; Found: 175.0010.
8.
Potassium
O-ethyl xanthate (99%) was obtained from Aldrich (listed under ethylxanthic acid potassium salt) and recrystallized from hot ethanol before use. The checkers used the commercial product as received.
9.
The progress of the reaction was monitored by TLC analysis on silica gel using hexanes/ethyl acetate (4:1) as eluent; the product had an R
f = 0.23 (stained with
p-anisaldehyde solution [(
950 mL of ethanol, 95%,
35 mL of concentrated sulfuric acid,
26 mL of p-anisaldehyde,
10.5 mL of acetic acid)].
10.
The product exhibited the following properties: mp 84–86 °C;
1H NMR
pdf (CDCl
3, 400 MHz) δ: 1.45 (t,
J = 7.0 Hz, 3 H), 2.11 (s, 3 H), 4.70 (q,
J = 7.0 Hz, 2 H), 6.61 (qd,
J = 7.6, 9.7 Hz, 1 H), 7.43 (d,
J = 9.7 Hz, 1 H);
13C NMR
pdf (CDCl
3, 100 MHz) δ: 13.6, 22.8, 57.9 (q,
J = 38 Hz), 71.4, 123.5 (q,
J = 279 Hz), 170.9, 207.1; IR (film): 3278, 2990, 2933, 1667, 1514, 1370, 1334, 1289, 1246, 1210, 1186, 1127, 1115, 1102, 1049, 857, 819, 684 cm
−1. The product obtained following the procedure was pure enough for use in the next step. However, it could be recrystallized by dissolving 1 g of the compound in a hot mixture of
1 mL of ethyl acetate and
10 mL of heptane and allowing the solution to cool to room temperature whereupon the product crystallized (yield of crystallization > 85%). Anal. Calcd for C
7H
10F
3NO
2S
2: C, 32.18; H, 3.86. Found: C, 32.57; H, 3.91.
11.
Vinyl acetate (99+%) and lauroyl peroxide (DLP, 97%) were obtained from Aldrich, and used as received. 1,2-Dichloroethane (99+%) was purchased from SDS Carlo Erba and used as received. The checkers used 1,2-dichloroethane (99%) purchased from KMF.
12.
When performed on a smaller scale (
4.28 g of xanthate 3) complete conversion was reached with only 3 portions of lauroyl peroxide with a reaction time of only 4.5 h.
13.
The progress of the reaction was followed by TLC analysis on silica gel using hexanes/ethyl acetate (4:1) as eluent and visualization was performed with
p-anisaldehyde solution. The product had an R
f = 0.075.
14.
The product exhibited the following properties:
1H NMR
pdf (CDCl
3, 400 MHz, mixture of diastereomers) δ: 1.30 (br. t, 3 H), 1.41 (t,
J = 7.0 Hz, 3 H), 2.02 (s, 6 H), 2.07 (s, 6 H), 2.10–2.27 (m, 2 H), 2.44–2.51 (m, 2 H), 4.61–4.67 (m, 4 H), 4.79–4.85 (m, 2 H), 6.54 (dd,
J = 3.0, 9.7 Hz, 2 H), 6.65 (m, 2 H); IR (CCl
4): 3429, 2982, 1767, 1706, 1503, 1369, 1235, 1188, 1137, 1049 cm
−1. MS (ESI): 370 ([M+Na]
+).
15.
(±)-10-Camphorsulfonic acid was purchased from Acros, 98%, and used as received.
16.
Approximately 150 g of silica (E. Merck, Darmstadt, 230–240 mesh) was used with hexanes/ethyl acetate (2:3) as the eluent, R
f = 0.16 (stained with potassium permanganate solution).
17.
The submitters reported that the crude product could be purified by crystallization by cooling a solution in Et
2O (20 mL) to −78 °C (52% yield). The checkers, however, experienced problems caused by gelation of the mixture and therefore utilized chromatographic purification.
18.
The product exhibited the following properties: mp 61–63 °C (ether).
1H NMR
pdf (CDCl
3, 400 MHz) δ: 1.83 (ddd,
J = 3.9, 9.8, 14.2 Hz, 1 H), 2.03 (s, 3 H), 2.04 (ddd,
J = 3.3, 7.6, 14.2 Hz, 1 H), 3.34 (s, 3 H), 3.36 (s, 3 H), 4.46 (dd,
J = 7.6, 3.9 Hz, 1 H), 4.75 (m, 1 H), 6.89 (d,
J = 9.6 Hz, 1 H);
13C NMR
pdf (CDCl
3, 100 MHz) δ: 22.9, 31.7, 47.6 (q,
J = 32 Hz), 53.1, 53.9, 101.6, 125.2 (q,
J = 280 Hz), 170.6; IR (CCl
4): 3285, 3072, 2939, 2834, 1665, 1551, 1437, 1375, 1299, 1265, 1181, 1135, 1064 cm
−1. MS (EI)
m/z (rel. intensity): 198 (45), 139 (58), 124 (56), 75 (100); HRMS (ESI): 252 ([M+Na]
+). Anal. Calcd for C
8H
14F
3NO
3: C, 41.92; H, 6.16. Found: C, 41.84; H, 6.09.
Handling and Disposal of Hazardous Chemicals
The procedures in this article are intended for use only by persons with prior training in experimental organic chemistry. All hazardous materials should be handled using the standard procedures for work with chemicals described in references such as "Prudent Practices in the Laboratory" (The National Academies Press, Washington, D.C., 2011 www.nap.edu). All chemical waste should be disposed of in accordance with local regulations. For general guidelines for the management of chemical waste, see Chapter 8 of Prudent Practices.
These procedures must be conducted at one's own risk. Organic Syntheses, Inc., its Editors, and its Board of Directors do not warrant or guarantee the safety of individuals using these procedures and hereby disclaim any liability for any injuries or damages claimed to have resulted from or related in any way to the procedures herein.
3. Discussion
The introduction of fluorine atoms in a given molecule often dramatically alters its chemical properties and its pharmacological profile in the case of a biologically active substance.
2 As a consequence, much ongoing effort has been devoted to the development of practical synthetic routes to the various classes of fluorinated compounds.
3 Radical reactions have proved quite useful in this respect. Xanthates, with their unique ability to mediate difficult radical processes such as intermolecular additions to non-activated olefins,
4 provide a collection of highly efficient reagents for the assembly of fluorinated derivatives. Some of these reagents are displayed in Figure 1. The transformation described above is representative of the use of xanthate
3.
5 Vinyl acetate can be replaced by a number of other olefinic traps, as shown by the examples in the Table (entries 1–5).
5 Xanthates
6 and
7 can be prepared by a modification of the route devised for the synthesis of compound
3.
6 They also add efficiently to various olefins as indicated by the examples in entries 6–10 in the Table.
6 A simple trifluoromethyl group can be introduced by the use of xanthate
8 (entry 11),
7 whereas reagent
9 gives directly a trifluoromethyl ketone (entry 12).
8
Figure 1: Reagents for the assembly of fluorinated derivatives
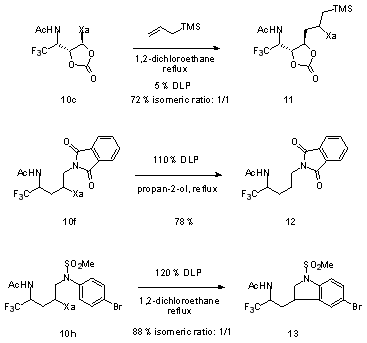
This approach to fluorinated derivatives combines efficiency with flexibility. The presence of the xanthate in the product can be exploited in many ways, since it provides an entry into the extremely rich chemistry of sulfur. It also allows the implementation of a second radical transformation, as shown by the three examples given in Scheme 1. The first example involves radical addition to allyl trimethylsilane leading to the densely functionalized structure
11.
5 The second transformation illustrates a process for the tin-free reductive removal of the xanthate group (
10f to
12).
6 The last example (
13) highlights the possibility of performing ring closures onto aromatic rings, providing a simple and direct entry into indolines.
6Table 1: Radical Additions of Xanthates (Xa = S-C(=S)OEt).
Scheme 1: Radical transformations of adducts (DLP = dilauroyl peroxide)
Appendix
Chemical Abstracts Nomenclature (Collective Index Number);
(Registry Number)
2,2,2-Trifluoro-1-methoxyethanol; (431-46-9)
Acetamide; (60-35-5)
N-(2,2,2-Trifluoro-1-hydroxyethyl)-acetamide; (6776-45-0)
Thionyl chloride; (7719-09-7)
N-1-(Chloro-2,2,2-trifluoroethyl)acetamide; (6776-46-1)
Potassium O-ethyl xanthate:
Carbonodithioic acid,
O-ethyl ester,
potassium salt; (140-89-6)
S-(1-Acetylamino-2,2,2-trifluoroethyl) O-ethyl dithiocarbonate:
Carbonodithioic acid,
S-[1-(acetylamino)-2,2,2-trifluoroethyl] O-ethyl ester; (583029-16-7)
Lauroyl peroxide:
Peroxide, bis(1-oxododecyl); (105-74-8)
Vinyl acetate:
Acetic acid ethenyl ester (108-05-4)
3-Acetylamino-1-ethoxythiocarbonylsulfanyl-4,4,4-trifluorobutyl acetate:
Carbonodithioic acid,
S-[3-(acetylamino)-1-(acetyloxy)-4,4,4-trifluorobutyl] O-ethyl ester: (583028-99-3)
(±)-10-Camphorsulfonic acid; (5872-08-2)
N-(3,3-Dimethoxy-1-trifluoromethyl-propyl)-acetamide:
Acetamide,
N-[3,3-dimethoxy-1-(trifluoromethyl)propyl]-; (583029-14-5)
 |
Samir Z. Zard was born in 1955 in Ife, Nigeria. His training as a chemist started at the American University of Beirut, then at Imperial College, London, and finally at the Université Paris-Sud, France, where he received his doctorate under the supervision of Professor Sir Derek Barton in 1983. His main research interests concern the study and development of new reactions and processes. |
 |
Fabien Gagosz graduated from the Chemistry Scool of Strasbourg in 1997. He obtained his PhD in 2002 from the Ecole Polytechnique in Palaiseau. He worked under the supervision of Prof. Samir Z. Zard on the development of new radical reactions with applications to the synthesis of alkaloids, phosphorylated and fluorinated compounds. He then joined Prof. W. B. Motherwell at the University college of London in 2003 as a postdoctoral associate. He returned to the Ecole Polytechnique in 2004 and joined the CNRS as Chargé de Recherches. His current independent research focuses on the development of new gold(I) catalysts and their use in the design of new synthetic methodologies. |
 |
Daniel Laurich was born in 1982 in Essen, Germany. After completing gymnasium (high school), he started his education in 1999 as a laboratory assistant at the Max-Planck-Institute for Coal Research in the group of Professor Alois Fürstner. He completed these studies in 2002 and remained in the group to work on various projects ranging from organometallic chemistry to natural product total synthesis. In 2003, he decided to further his experimental training by beginning studies to become a laboratory technician. |
Copyright © 1921-, Organic Syntheses, Inc. All Rights Reserved