Org. Synth. 2013, 90, 306-315
DOI: 10.15227/orgsyn.090.0306
Low-epimerization Peptide Bond Formation with Oxyma Pure: Preparation of Z-L-Phg-Val-OMe
Submitted by Ramon Subirós-Funosas,
* Ayman El-Faham
*, and Fernando Albericio.
*
1
Checked by Asher Lower and Margaret Faul.
1. Procedure
Z-L-Phg-Val-OMe. A 500-mL, three-necked, round-bottomed flask equipped with a nitrogen gas inlet, thermocouple and Teflon-coated cylindrical magnetic stirrer bar (50 x 8 mm) (Note 1), is charged with EDC-HCl (4.72 g, 25 mmol, 1 equiv) and dissolved in DCM/DMF (1:1) (110 mL) ( Notes 2 and 3). The colorless solution of the carbodiimide is then stirred (700 rpm) at room temperature for approximately 20 min to allow dissolution, followed by immersion in an ice bath at 0 °C.
Z-L-Phg-OH (7.26 g, 25 mmol, 1 equiv) and Oxyma Pure (3.67 g, 25 mmol, 1 equiv) are added to the cold solution of EDC-HCl as solids (Notes 4, 5 and 6). Two minutes after the addition of the reactants, H-Val-OMe-HCl (4.27 g, 25 mmol, 1 equiv) is added as a solid to the preactivated cocktail, followed by addition of DIEA (4.29 mL, 25 mmol, 1 equiv) using a 5-mL syringe (Note 7). The flask is then flushed with nitrogen and sealed with a polyethylene cap. The coupling cocktail is allowed to stir at 700 rpm in the ice bath for 1 h and then the reaction progresses at room temperature overnight. After 14-15 hours, the extent of peptide bond formation is monitored by TLC, showing trace amounts of starting acid and amine (Note 8). The solvent is removed by rotary evaporation (40 °C, 28 mmHg) (Note 9). The crude oily residue is diluted in AcOEt (500 mL) and transferred to a 1-L graduated separatory funnel (Note 10). The organic solution is extracted and washed with 1 N HCl (3 x 300 mL), 1 N Na2CO3 (3 x 350 mL), and saturated NaCl (3 x 350 mL). The resulting pale yellow organic fraction is dried over approximately 25 g of anhydrous MgSO4, filtered to a 1-L round-bottomed flask, and concentrated by rotary evaporation (25 °C, 28 mmHg) to afford a white solid, with partial yellow tone, which is dried overnight in a vacuum oven (50 °C, =10 mmHg) to yield crude product (9.57 g, 97.5% yield) (Note 11). Ethanol (100 mL) is added to the peptidic material at room temperature and the resulting slurry is stirred at 200 rpm on the rotary evaporator. After complete dissolution of the solid at 60 °C bath temperature, the stirring and heating of the solution is stopped and the solution is allowed to cool slowly to room temperature over 2 h, resulting in crystallization of the dipeptide as white needles within the yellow solution. The resulting needles are isolated by filtration through a Büchner funnel, washed with ice-cold ethanol (100 mL), collected in a 25 x 100 mm crystallizing dish and dried overnight in a vacuum oven (50°C, 3 mmHg) to provide Z-L-Phg-Val-OMe dipeptide (7.98-8.25 g, 81-84% yield) as white needles, free of the DL-epimer (Note 12).
2. Notes
1. In order to enhance the efficiency of the coupling process, all glassware was dried overnight in an oven at 70 °C and flushed with a stream of
nitrogen after cooling to prevent the presence of air or moisture.
2. Anhydrous
dichloromethane (
DCM) (99.8%, stabilized with
amylene) and
N,N-dimethylformamide (
DMF, anhydrous, 99.8%) were purchased from Sigma Aldrich.
Z-L-Phg-OH (99.5%) was obtained from Iris Biotech.
H-Val-OMe hydrochloride (99%) was purchased from Sigma Aldrich.
1-Ethyl-3-(3-dimethylaminopropyl)carbodiimide hydrochloride (
EDC-HCl) (premium quality, crystalline) was purchased from Fisher Scientific.
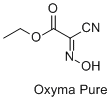
Ethyl 2-cyano-2-hydroxyimino acetate (
Oxyma Pure) (99.9%) was obtained from Bachem.
N,N-Ethyldiisopropylamine (
DIEA) (99.5%) was purchased from Fisher Scientific. All chemicals were used as received and stored at room temperature on the bench top, except for the amino acids and
EDC-HCl, which were stored in a freezer at -5°C given their hygroscopic nature.
3. New bottles of
DCM and
DMF packaged in 100-mL Sure-Seal amber bottles were used.
4. Addition of the reagents should take place no more than 5 minutes after the cooling of the
carbodiimide solution, since
EDC-HCl tends to precipitate at 0 °C. Following addition of
Z-L-Phg-OH and
Oxyma Pure any insoluble
EDC-HCl quickly dissolves.
5.
Z-L-Phg-OH and
Oxyma Pure are preferably weighed together and added as a solid mixture for an optimized coupling.
6. Immediately after contacting the acid and
Oxyma Pure, the
carbodiimide solution turned from colorless to bright yellow.
7. After addition of the amine and base, the solution acquired a bright orange color, which slowly decreased with reaction progress to pale yellow.
8. TLC analyses were performed using plates pre-coated with silica Gel 60 F254, purchased from EMD Chemicals Inc., using 10%
DCM/
MeOH with 1%
AcOH as eluent. The R
f value of the title dipeptide product is 0.73, whereas the starting
Z-L-Phg-OH and
H-Val-OMe-HCl have R
f values of 0.41 and 0.14 respectively. Phosphomolybdate dip solution allowed clear visualization of
Z-L-Phg-OH and
Z-L-Phg-Val-OMe in the crude mixture, whereas
H-Val-OMe-HCl (bright red) could be clearly spotted with alternative ninhydrin staining.
9.
DMF was co-evaporated at 40 °C with approximately 200 mL of
toluene.
10. Although the dipeptide product is more easily dissolved in
dichloromethane, more facile extraction and greater yields are obtained with this methodology.
11. A small sample (10 mg) of the crude mixture obtained after work-up is analyzed by
1H NMR,
pdf showing approximately 1.6% of the DL-epimer The most reliable and accurate method to determine the extent of epimerization in dipeptides containing
N-terminal Phg residues is the integration of methoxy peaks corresponding to the different epimers in the
1H NMR spectra, taking advantage of the methyl ester nonequivalence.
2With regard to the dipeptide herein described, LL and DL epimers displayed OMe signals at δ 3.63 and 3.73 ppm, respectively, in CDCl
3.
3 The integration method described by Davies can be applied, and the result is in good agreement with simple normalization of the methyl resonance.
4 The limit of detection for NMR is considered =0.2% and limit of quantification is =0.5%. Analysis of the degree of epimerization was also attempted by reverse-phase HPLC, but complete separation of the LL and DL epimers could not be achieved.
12. Diastereomeric purity of the recrystallized solid was again examined by
1H NMR spectroscopy, showing complete disappearance of the OMe signal of the DL-epimer at δ 3.73 ppm. The recrystallized material showed also excellent overall purity (>99%) according to HPLC and NMR techniques. The submitter reported the following HPLC conditions could be used to assess purity. HPLC was conducted on a X-Bridge BEH column (C
18 3.5 mm, 4.6 x 100 mm), using a linear gradient of 5 to 100%
CH3CN in
H2O/0.1%
TFA over 8 min, with a flow rate 1.0 mL/min and detection at 220 nm, Retention timeof (
Z-L-Phg-Val-OMe) = 7.25 min. The
Z-L-Phg-Val-OMe obtained using this procedure, which is stable after long storage in a freezer at -5 °C, has the following physical and spectral data: mp: 147-148 °C; FTIR (neat, cm
-1): 3338 (m), 3281 (s), 3061 (w), 3033 (w), 2967 (w), 2874 (w), 1740 (s), 1706 (m), 1659 (s), 1531 (s), 1497 (w), 1456 (w), 1433 (w), 1390 (w), 1363 (m), 1341 (w), 1305 (w), 1278 (w), 1250 (m), 1213 (m), 1141 (m), 1114 (w), 1079 (w), 1050 (w), 1028 (w), 1012 (w), 964 (w), 915 (w), 898 (w), 753 (m), 733 (m), 699 (s), 629 (s).
1H-NMR
pdf(400 MHz, CDCl
3) δ: 0.86 (d,
J = 6.7 Hz, 3 H, CH
3), 0.92 (d,
J = 6.9 Hz, 3 H, CH
3), 2.15 (m, 1 H, CH), 3.63 (s, 3 H, OCH
3), 4.49 (dd,
J = 4.9, 8.6 Hz, 1 H), 5.09 (d,
J = 12.3 Hz, 2 H, OCH
2), 5.27 (d,
J = 3.6 Hz, 1 H, NH), 6.06 (d,
J = 7.4 Hz, 1 H, NH), 6.18 (d,
J = 8.1 Hz, 1 H, CH), 7.27-7.40 (m, 10 H, CH
ar);
13C NMR
pdf(100 MHz, CDCl
3) δ: 17.8, 18.9, 31.3, 52.1, 57.5, 59.0, 67.1, 127.3, 128.1, 128.1, 128.5, 128.7, 129.1, 136.2, 137.5, 155.7, 169.7, 171.7; [α]
20D= +77.4 (CHCl
3, c = 1.00); HRMS (
m/z) (ESI): calcd. for C
21H
27N
2O
5 [M+H] 399.19145, found 399.19124; TLC (Hexane/
AcOEt = 1:1) R
f = 0.69;
Handling and Disposal of Hazardous Chemicals
The procedures in this article are intended for use only by persons with prior training in experimental organic chemistry. All hazardous materials should be handled using the standard procedures for work with chemicals described in references such as "Prudent Practices in the Laboratory" (The National Academies Press, Washington, D.C., 2011 www.nap.edu). All chemical waste should be disposed of in accordance with local regulations. For general guidelines for the management of chemical waste, see Chapter 8 of Prudent Practices.
These procedures must be conducted at one's own risk. Organic Syntheses, Inc., its Editors, and its Board of Directors do not warrant or guarantee the safety of individuals using these procedures and hereby disclaim any liability for any injuries or damages claimed to have resulted from or related in any way to the procedures herein.
3. Discussion
Peptides have emerged in the last decade as promising therapeutic scaffolds in the pharmaceutical industry, given their high potency, specificity and low toxicity.
5 Retention of
optical purity throughout the chemical process is pivotal for these New Chemical Entities and consequently, methodological tools have been developed to minimize C
α-epimerization.
6, 7 In this regard, the inclusion of
N-hydroxylamines in the coupling cocktail helps prevent the loss of stereochemical integrity in the activated intermediate species.
8 The
N-hydroxybenzotriazole family, which comprises
HOBt,
HOAt and
6-Cl-HOBt as the most renowned members, have dominated the field in the preceding decades over succinimides (HOSu), benzotriazines (HODhb), pyridinones (HOPy) and triazoles (HOCt), as a result of their enhanced reactivity, absence of side reactions, and overall moderate prices.
7 However, the dangerous safety profile of
N-hydroxybenzotriazoles and derived coupling reagents prompted a search for alternative templates.
7, 8, 9 Recently, acidic ketoximes featuring electron-withdrawing substituents have been proposed as additives to carbodiimides, leading to the widespread reception of ethyl 2-cyano-2-hydroxyimino acetate (
Oxyma Pure) as a reliable and safe coupling choice.
7, 8, 10 The ability of
Oxyma Pure to preserve stereochemical configuration during activation of peptide fragments and Cys proved much higher than that of
HOBt and comparable to that of
HOAt.
10
Best results have been obtained during activation of the highly sensitive
α-phenylglycine (Phg) residue towards epimerization, with
Oxyma Pure exceeding the performance of
HOAt in minimizing the level of Z-
D-Phg-Pro-NH
2 (Table 1).
2, 11 Moreover, the use of
Oxyma Pure in combination with formamidinium salts results in further reduction of epimerization.
12 In addition,
Oxyma Pure provides considerably broader scope than
HOBt by mediating difficult junctions, such as those containing
N-Me or Aib residues, and is fully compatible with microwave irradiation.
10 Recently, other applications such as ester and amide bond formation, and the minimization of base-catalyzed side reactions, have been discovered.
13, 14
This procedure describes the application of
Oxyma Pure in the preparation of the
Z-L-Phg-Val-OMe dipeptide, which combines the epimerization-prone α-phenylglycine amino acid and the sterically hindered valine residue.
3, 15, 16 Therefore, this dipeptide (which has been reported a few times in the literature) is an excellent platform to test both the capability of
Oxyma Pure to retain stereochemical purity and to promote acylation of bulky residues.
3, 15, 16 An advantage of this peptide model over other Phg-containing dipeptides such as Z-Phg-Pro-NH
2 is the ability to monitor the degree of epimerization by
1H-NMR (see
Note 11) and the ease of spectral analysis, given the absence of cis/trans isomerism.
2, 3, 16 In addition,
EDC-HCl is employed as
carbodiimide because of its more facile removal than DIC during aqueous work-up. Although the basic center contained in its structure could promote epimerization, this effect is not observed in solution-phase; moreover,
EDC-HCl accelerates active ester formation to a greater extent than DIC.
17 A similar enhancement in the activation process is observed when
DCM is used as coupling solvent, with simultaneous reduction of loss of chirality.
11, 17 Using this combination of reagents and solvents, an optimized method for
Oxyma Pure-mediated peptide bond formation is presented. All reagents are commercially available at low to moderate cost and furthermore,
Oxyma Pure is comparatively less expensive than
HOAt.
By means of the abovementioned procedure, the suitability of the
EDC-HCl/
Oxyma Pure coupling system in solution-phase is unambiguously demonstrated (Table 2). In comparison to previous procedures on the same peptide platform using
N-hydroxybenzotriazoles,
Oxyma Pure afforded a substantially lower content of DL epimer than
HOBt (0.1 vs 3.7%) with concomitant increase of yield.
15 The extent of epimerization was also considerably lower than that induced by
HOAt (0.1 vs. <1-2%).
15 Similar to the tendency observed with the
N-hydroxylamines,
Oxyma Pure also outperformed the corresponding benzotriazole-based aminium salts HATU and HBTU, and formamidinium salt
TFFH, even when the low epimerization-inducing
DCM is used as solvent (Table 2).
3, 15, 16 Further, not only yield and stereochemical integrity, but also purity, is enhanced with
Oxyma Pure, based on the melting point of the target dipeptide obtained using the present (147-148°C) vs. previous (136-141°C) procedures.
3, 16 Moreover, the procedure showed that after recrystallization the level of the epimer is negligible.
Table 1. Extent of epimerization during stepwise formation of Z-L-Phg-Pro-NH2 in solution phase.
 a
a 2 min. preactivation was performed with 2 equiv of
DIEA.
Table 2. Extent of epimerization during stepwise formation of Z-L-Phg-Val-OMe in solution phase.
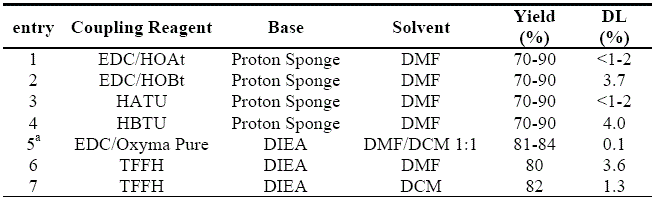 a
a This work, after recrystallization
Appendix
Chemical Abstracts Nomenclature; (Registry Number)
Z-L-Phg-OH: Benzeneacetic acid, α-[[(phenylmethoxy)carbonyl]amino]-, (αS)- (1) (53990-33-3)
H-Val-OMe-HCl: L-Valine, methyl ester, hydrochloride (1:1) (2) (6306-52-1)
Z-L-Phg-Val-OMe: L-Valine, (2S)-2-phenyl-N-[(phenylmethoxy)carbonyl]glycyl-, methyl ester (3) (159487-14-6)
EDC-HCl: 1,3-Propanediamine, N3-(ethylcarbonimidoyl)-N1,N1-dimethyl-, hydrochloride (1:1) (4) (25952-53-8)
Oxyma Pure: Acetic acid, 2-cyano-2-(hydroxyimino)-, ethyl ester (5) (3849-21-6)
DIEA: 2-Propanamine, N-ethyl-N-(1-methylethyl)- (6) (7087-68-5)
DCM: Methane, dichloro- (7) (75-09-2)
DMF: Formamide, N,N-dimethyl- (8) (68-12-2)

|
Professor Ayman El-Faham received his B.Sc. and M.Sc. in Physical Organic Chemistry from the University of Alexandria, Egypt. In 1991 he received his Ph.D. in organic chemistry through a joint project between the University of Alexandria and the University of Massachusetts. From 1992 to 1999, he worked on new coupling reagents at Professor Carpino's Lab. Following a position as Head of the Chemistry Department, Beirut Arab University (2000 to 2004), and as Direct Manager of both the NMR and Central Lab at Alexandria University (2004 to 2008), he worked at King Saud University, as a Professor of Organic Chemistry (2008 to 2010). Currently he is working back at King Saud University. His research interests include synthesis of peptides, natural products, heterocycles, and biologically active targets.
|

|
Ramon Subirós-Funosas received his B.Sc. in Chemistry from the University of Barcelona in 2007. Previously, he moved for one year to GlaxoSmithKline, in Stevenage, UK, where he joined the Medicinal Chemistry Department in 2005. Ramon obtained his Ph.D. in Organic Chemistry in 2011 from the University of Barcelona, under the supervision of Prof. Fernando Albericio, developing a new family of coupling reagents based on Oxyma Pure. In 2010, he visited the group of Professor Dawson at The Scripps Research Institute, La Jolla, USA, for a 4-month internship, acquiring knowledge in the synthesis of small proteins. His major research interests include peptide synthesis and methodology of native chemical ligation. He is currently a Marie Curie IEF fellow at Humboldt Universität-zu-Berlin with Prof. Oliver Seitz.
|

|
Professor Fernando Albericio obtained his Ph.D. in Chemistry from the University of Barcelona. After postdoctoral work at Tufts University, at the Université d'Aix-Marseille, and at the University of Minnesota, he joined the University of Barcelona as an Associate Professor. In 1992-1994, he was appointed Director of Peptide Research at Milligen/Biosearch, Boston, USA, then returned to the University of Barcelona. Currently, he is holding a triple appointment as Professor at the University of Barcelona and Group Leader at the Institute for Research in Biomedicine, and Research Professor at the University of KwaZuluNatal (Durban, South Africa). From 2005 to 2012, he has been Executive Director in the Barcelona Science Park. His major research interests cover practically all aspects of peptide synthesis and combinatorial chemistry methodology. |

|
Asher Lower was born in San Francisco, CA in 1981. He received his Ph.D. in synthetic organic chemistry from the University of California, Santa Barbara in 2006 under the direction of Bruce H. Lipshutz. His doctoral research focused on the development of new stereoselective transformations using inexpensive and environmentially friendly Cu and Ni catalysts. Asher started his career at Ampac Fine Chemicals where he held the position of Sr. Scientist specializing in commercial-scale manufacture of hazardous APIs. Asher joined Amgen in 2009 and currently supports late-stage and commercial process development.
|
Copyright © 1921-, Organic Syntheses, Inc. All Rights Reserved