Org. Synth. 1972, 52, 39
DOI: 10.15227/orgsyn.052.0039
THE FORMATION AND ALKYLATION OF SPECIFIC ENOLATE ANIONS FROM AN UNSYMMETRICAL KETONE: 2-BENZYL-2-METHYLCYCLOHEXANONE AND 2-BENZYL-6-METHYLCYCLOHEXANONE
[Cyclohexanone, 2-methyl-2-(phenylmethyl)- and cyclohexanone, 2-methyl-6-(phenylmethyl)-]
Submitted by Martin Gall and Herbert O. House
1.
Checked by K. E. Wilson and S. Masamune.
1. Procedure
A. 2-Methyl-1-cyclohexen-1-yl acetate. Caution! Since mixtures of perchloric acid with small amounts of organic material can explode violently, the perchloric acid should always be the last component added to the reaction mixture.
To a 1-l. flask are added 600 ml. of carbon tetrachloride, 270 g. (250 ml., 2.65 moles) of acetic anhydride, 56 g. (0.50 mole) of 2-methylcyclohexanone, and 0.34 ml. (0.002 mole) of 70% perchloric acid. The reaction flask is stoppered and allowed to stand at room temperature for 3 hours during which time the reaction solution becomes first yellow-orange and finally red in color. The reaction mixture is poured into a cold (0–5°) mixture of 400 ml. of saturated aqueous sodium hydrogen carbonate and 400 ml. of pentane contained in a 4-l. Erlenmeyer flask equipped with a mechanical stirrer. While the mixture is stirred vigorously at 0–5°, solid sodium hydrogen carbonate is added in 3–5 g. portions as rapidly as foaming of the reaction mixture will permit. The addition of solid sodium hydrogen carbonate is continued until the acetic acid has been neutralized and the aqueous phase remains slightly basic (pH 8). This neutralization requires approximately 400 g. of solid sodium hydrogen carbonate added in portions over a period of ca. 3 hours. As soon as the neutralization is complete (Note 1), the organic layer (the lower layer) is separated and the aqueous phase is extracted with three 200-ml. portions of pentane. The combined organic solutions are dried over anhydrous magnesium sulfate and concentrated by distilling the bulk of the pentane through a 30-cm. Vigreux column. The remaining solvents are removed with a rotary evaporator and the residual liquid is distilled (Note 2) under reduced pressure, yielding 66.6–70.9 g. (87–92%) of 2-methyl-1-cyclohexen-1-yl acetate, as a colorless liquid, b.p. 81–86° (18 mm.), nD25 1.4562–1.4572 (Note 3).
B. 2-Benzyl-2-methylcyclohexanone. Caution! Ethereal solutions of methyllithium in contact with atmospheric oxygen may catch fire spontaneously. Therefore any manipulations with this reagent must be carried out with the utmost care to avoid accidental spillage. Benzyl bromide is a powerful lachrymator. Steps B and C should be performed in an efficient fume hood.
A 1-l., three-necked flask is equipped with a nitrogen-inlet tube fitted with a stopcock, a glass joint fitted with a rubber septum, a 125-ml., pressure-equalizing dropping funnel, a thermometer, and a glass-covered magnetic stirring bar. After the apparatus has been dried in an oven, 20 mg. of 2,2'-bipyridyl is added to the flask and the apparatus is thoroughly flushed with anhydrous, oxygen-free nitrogen (Note 4). A static nitrogen atmosphere is maintained in the reaction vessel throughout subsequent operations involving organometallic reagents (Note 5). An ethereal solution containing 0.40 mole of methyllithium (Note 6) is added to the reaction vessel with a hypodermic syringe. The diethyl ether is removed by evacuating the apparatus while the solution is stirred and the flask is warmed with a water bath (40°) (Note 7). The reaction vessel is refilled with nitrogen and 400 ml. of 1,2-dimethoxyethane (b.p. 83°, freshly distilled from lithium aluminum hydride) is transferred to the reaction vessel with a hypodermic syringe or a stainless steel cannula. The resulting purple solution of methyllithium and the methyllithium bipyridyl charge-transfer complex is cooled to 0–10° before 29.3 g. (0.190 mole) of 2-methyl-1-cyclohexen-1-yl acetate is added, dropwise and with stirring, over a period of 35–45 minutes (Note 8) while the temperature of the reaction mixture is maintained at 0–10° with an ice bath. After the addition of the enol acetate, the reaction solution must still retain a light red-orange color indicating the presence of a small amount of excess methyllithium (Note 9). To this cold (10°) solution is added rapidly (10–15 seconds) and with stirring, 68.4 g. (0.400 mole) of freshly distilled benzyl bromide [b.p. 78–79° (12 mm.), nD25 1.5738]. The resulting yellow solution is stirred for 2–2.5 minutes (during which time the temperature of the reaction mixture rises from 10° to about 30°), poured into 500 ml. of cold (0–10°), saturated aqueous sodium hydrogen carbonate, and extracted with three 150-ml. portions of pentane. The combined organic extracts are dried over anhydrous magnesium sulfate and concentrated with a rotary evaporator. The residual liquid is fractionally distilled under reduced pressure, separating 31–41 g. of forerun fractions, b.p. 71–89° (20 mm.) and 41–87° (0.3 mm.) (Note 10), and 20.7–22.2 g. (54–58%) of 2-benzyl-2-methylcyclohexanone as a colorless to pale yellow liquid, b.p. 87–93° (0.3 mm.), nD25 1.5322–1.5344 (Note 11) and (Note 12).
C. 2-Benzyl-6-methylcyclohexanone. Caution! The same precaution as that described in part B should be exercised in this step.
A 1-l., three-necked flask is equipped as described in part B. After the assembled apparatus has been dried in an oven, 45 mg. of 2,2'-bipyridyl is added to the flask and the apparatus is thoroughly flushed with anhydrous, oxygen-free nitrogen (Note 4). A static nitrogen atmosphere is maintained in the reaction vessel throughout subsequent operations involving organometallic reagents (Note 5). An ethereal solution containing 0.20 mole of methyllithium (Note 6) is added to the reaction flask with a hypodermic syringe. After the ether is removed under reduced pressure as described in part B (Note 7), the reaction vessel is refilled with nitrogen and 400 ml. of 1,2-dimethoxyethane (b.p. 83°, freshly distilled from lithium aluminum hydride) is added to the vessel with a hypodermic syringe or a stainless steel cannula. The resulting purple solution of methyllithium and the methyllithium-bipyridyl charge-transfer complex is cooled to −50° with a dry ice–methanol bath before 21.0 g. (29.2 ml., 0.208 mole) of diisopropylamine (b.p. 84–85°, freshly distilled from calcium hydride) is added with a hypodermic syringe, dropwise and with stirring. During this addition, which requires 2–3 minutes, the temperature of the reaction solution should not be allowed to rise above −20° (Note 12). The resulting reddish-purple solution of lithium diisopropylamide and the bipyridyl charge-transfer complex is stirred at −20° for 2–3 minutes before 50 ml. of a 1,2-dimethoxyethane solution containing 21.3 g. (0.190 mole) of 2-methylcyclohexanone is added, dropwise and with stirring. During this addition the temperature of the reaction solution should not be allowed to rise above 0° (Note 12). After the addition of the ketone, the solution of the lithium enolate must still retain a pale reddish-purple color indicating the presence of a slight excess of lithium diisopropylamide (Note 9) and (Note 13). The enolate solution is stirred and warmed to 30° with a water bath before 68.4 g. (0.400 mole) of freshly distilled benzyl bromide [b.p. 78–79° (12 mm.), nD25 1.5738] is added, rapidly and with vigorous stirring, from a hypodermic syringe. The temperature of the reaction mixture rises to about 50° within 2 minutes and then begins to fall. After a total reaction period of 6 minutes, the reaction mixture is poured into 500 ml. of cold (0–10°), saturated aqueous sodium hydrogen carbonate and extracted with three 150-ml. portions of pentane. The combined organic extracts are washed successively with two 100-ml. portions of 5% hydrochloric acid and 100 ml. of saturated aqueous sodium hydrogen carbonate, dried over anhydrous magnesium sulfate, and concentrated with a rotary evaporator. The residual yellow liquid is fractionally distilled under reduced pressure (Note 14), separating 31–32 g. of forerun fractions, b.p. 67–92° (20 mm.) and 40–91° (0.3 mm.) (Note 10), and 21.3–23.3 g. (58–61%) of crude 2-benzyl-6-methylcyclohexanone as a colorless liquid, b.p. 91–97° (0.3 mm.), nD25 1.5282–1.5360. The residue (10–11 g.) contains dibenzylated products. The crude reaction product contains (Note 11) and (Note 15) 2-benzyl-6-methylcyclohexanone (86–90%) and 2-benzyl-2-methylcyclohexanone (10–14%) accompanied in some cases by small amounts of trans-stilbene (Note 13).
To obtain the pure 2,6-isomer, the following procedure may be followed. A 200-ml. flask, equipped with a Teflon-covered magnetic stirring bar, dried in an oven, and flushed with nitrogen, is charged with 2.59 g. (0.0479 mole) of sodium methoxide (Note 16) and stoppered with a rubber septum. A static nitrogen atmosphere is maintained in the reaction vessel throughout the remainder of the reaction. Using a hypodermic syringe, 90 ml. of ether (freshly distilled from lithium aluminum hydride) is added to the flask. The resulting suspension is cooled with an ice bath before a mixture of the crude, distilled, alkylated product (about 21–23 g.) and 3.74 g. (0.0505 mole) of ethyl formate (Note 17) is added with a hypodermic syringe. The mixture is stirred for 10 minutes with ice-bath cooling. The bath is then removed and stirring is continued for an additional 50 minutes. The resulting yellow suspension is treated with 300 ml. of water and extracted with 250 ml. of ether. The ethereal extract is washed with 100 ml. of aqueous 1 M sodium hydroxide, dried over anhydrous magnesium sulfate, and concentrated with a rotary evaporator. The residual yellow liquid is distilled under reduced pressure, yielding 16.2–17.3 g. (overall yield 42–45%) of pure (Note 11) 2-benzyl-6-methylcyclohexanone as a colorless liquid, b.p. 95–100° (0.3 mm.), nD25 1.5299–1.5328.
2. Notes
1.
Because the enol acetate is slowly hydrolyzed, even by neutral aqueous solutions, the reaction mixture should be neutralized and the organic product separated and dried as rapidly as is practical.
2.
The glassware employed in the distillation should be washed first with
ammonium hydroxide, then water, and dried in an oven before use to avoid the possibility of acid-catalyzed hydrolysis or rearrangement of the enol acetate during the distillation.
3.
The submitters have been unsuccessful in finding a convenient
GC column which will separate
2-methyl-1-cyclohexen-1-yl acetate from its double-bond isomer,
6-methyl-1-cyclohexen-1-yl acetate. However, the
1H NMR spectrum (CCl
4) of the product exhibits a peak at δ 2.02 (singlet, C
H3CO) superimposed on a multiplet at δ 1.3–2.2 (vinyl C
H3 and aliphatic C
H2) and lacks absorption δ at 0.98 where
6-methyl-1-cyclohexen-1-yl acetate exhibits a doublet (
J = 7 Hz.) attributable to the aliphatic methyl group.
2 Consequently, the product contains less than 5% of the unwanted double-bond isomer. The product exhibits IR absorption (CCl
4) at 1755 cm.
−1 (enol ester C=O) and 1705 cm.
−1 (C=C).
4.
A
good grade of commercial, prepurified nitrogen can be used without further purification. A suitable method for the purification of
nitrogen is described in the literature.
35.
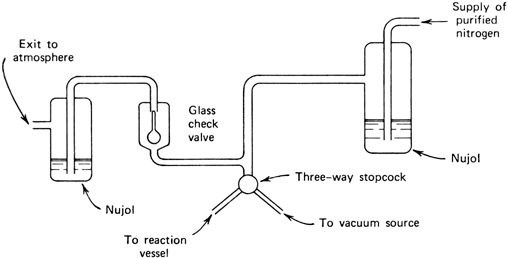
The apparatus illustrated in
Figure 1 is convenient both for evacuating the reaction vessel, refilling it with
nitrogen, and also for maintaining a static atmosphere of
nitrogen at slightly above atmospheric pressure in the reaction vessel.
Figure 1. Apparatus for either evacuating or supplying a nitrogen atmosphere to the reaction vessel.
6.
A solution of
methyllithium in ether was purchased from Alfa Inorganics, Inc. Directions for the preparation of
methyllithium from
methyl bromide are also available.
4 Solutions of
methyllithium should be standardized immediately before use by the titration procedure of Watson and Eastham.
5 A standard 0.500
M solution of
2-butanol (b.p.
99–100°, freshly distilled from
calcium hydride)
6 in
p-xylene (b.p.
137–138°, freshly distilled from
sodium) is prepared in a volumetric flask. A
25-ml. round-bottom flask, fitted with a rubber septum and a
glass-covered magnetic stirring bar, is dried in an oven. After
1–2 mg. of 2,2'-bipyridyl has been added to the flask, it is flushed with anhydrous, oxygen-free
nitrogen by inserting hypodermic needles through the rubber septum to allow gas to enter and escape. The tip of a
10-ml. burette is forced through the rubber septum and a measured volume of the standard
2-butanol solution is added to the flask, followed by
2.50 ml. of methyllithium solution. The mixture is stirred and additional standard
2-butanol solution is added to the flask from the burette until the purple color of the methyllithium-bipyridyl complex is just discharged. For a 1.66
M solution of
methyllithium,
8.30 ml. of the standard 2-butanol solution is required in this titration.
7.
Since some lithium enolates are significantly less soluble and less reactive in ether than in
1,2-dimethoxyethane, the submitters recommend the general use of this simple procedure to remove the ether before the lithium enolate is generated.
8.
Lithium enolates react relatively slowly with enol acetates to form
C-acetylated products. Consequently, the enol acetate should be added slowly with efficient stirring so that high local concentrations of both the enolate anion and the enol acetate are avoided.
9.
It is important that the indicator color, showing a small excess of strong base, not be discharged completely since the presence of any excess enol acetate or ketone will permit equilibration of the isomeric metal enolates. Consequently, the addition of this reactant is complete if further additions will discharge completely the color of the indicator.
10.
The various fractions of the forerun were analyzed employing a
GC column packed with silicone gum, No. XE-60, suspended on Chromosorb P and heated to 248°. The components found (retention times) were:
benzyl bromide (9.0 minutes),
2-methylcyclohexanone (5.3 minutes), and, in some cases,
bibenzyl (22.6 minutes). The
bibenzyl, formed by reaction of the
benzyl bromide with the excess
methyllithium7 was identified from the IR spectrum of a sample collected from the GC.
11.
Using a 6-m. GC column packed with silicone gum, No. XE-60, suspended on Chromosorb P and heated to 240°,
2-benzyl-2-methylcyclohexanone (retention time 35.0 minutes) and
2-benzyl-6-methylcyclohexanone (retention time 33.2 minutes,
cis- and
trans-isomers not resolved) are partially resolved. However, the use of this analytical method to detect small amounts of one structural isomer in the presence of the other is not reliable. GC, however, can be used to determine the presence of any
trans-stilbene (retention time 39.0 minutes) in the crude product.
The proportions of structurally isomeric benzylmethylcyclohexanones can be more accurately measured from the 1H NMR spectra of the distilled monoalkylated products. Pure 2-benzyl-6-methylcyclohexanone (principally the more stable cis-isomer in which both substituents are equatorial) exhibits the following 1H NMR (C6D6): δ 0.97 (d, J = 6.0 Hz., 3H, CH3), 1.1–2.6 (m, 9H, aliphatic CH and one of the two nonequivalent benzylic protons), 2.9–3.5 (m with at least 5 lines, 1H, the second of the nonequivalent benzylic protons), and 7.0–7.3 (m, 5H, aryl CH). In CCl4 the corresponding peaks are found at δ 0.97 (d, J = 6.0 Hz.), 1.1–2.7, 2.9–3.5, and 7.0–7.3; in this solvent a second weak doublet (J = 6.5 Hz.) is present at δ 1.04 and is attributable to the small amount of the less stable trans-2-benzyl-6-methylcyclohexanone (one equatorial to one axial substituent) present. The 2,6-isomer exhibits IR absorption (CCl4) at 1710 cm.−1 (C=O) and a series of weak (ε 202 to 335) UV maxima (95% C2H5OH) in the region 240–270 nm. The mass spectrum exhibits a molecular ion at m/e 202 with relatively abundant fragment peaks at m/e 159, 145, 117, 111, and 91 (base peak). Pure 2-benzyl-2-methylcyclohexanone has the following 1H NMR absorptions (C6D6): δ 0.91 (s, 3H, CH3), 1.2–1.7 (m, 6H, aliphatic CH), 2.1–2.4 (m, 2H, CH2CO), 2.78 (s, 2H, benzylic CH2), and 6.9–7.3 (m, 5H, aryl CH). In CCl4 the corresponding peaks are found at 0.95, 1.4–2.0, 2.2–2.6, 2.78, and 6.9–7.3. This ketone has IR absorption (CCl4) at 1710 cm.−1 (C=O) and shows a series of weak (ε 140 to 284) UV maxima (95% C2H5OH) in the region 240–270 nm. The mass spectrum exhibits a molecular ion at m/e 202 with relatively abundant fragment peaks at m/e 159, 117, 92, 91 (base peak), 55, 44, 43, and 41. Mixtures of the 2,6- and 2,2-isomer, could be analyzed by measuring their 1H NMR spectra in C6D6 and integrating the region δ 2.6–3.5. The peak at δ 2.78, attributable to both benzylic hydrogen atoms of the 2,2-isomer, is well resolved from the multiplet at δ 2.9–3.5, attributable to one of the two benzylic hydrogen atoms of the 2,6-isomer. Utilizing this method (which is in agreement with the less reliable value obtained by GC analysis), no 2,6-isomer is detected in the 2-benzyl-2-methylcyclohexanone prepared by the present procedure. The 2-benzyl-6-methylcyclohexanone product contains 10–14% of the 2,2-isomer.
12.
At temperatures above 0°,
1,2-dimethoxyethane is slowly attacked by
lithium diisopropylamide resulting in the protonation of the strong base.
13.
If this precaution is not followed, partial or complete equilibration of the enolates will occur because of proton transfers between the enolates and the excess un-ionized ketone. In an experiment where a slight excess of ketone was added, the distilled, monoalkylated product (40% yield) contained
77% of the undesired 2,2-isomer and only
23% of the desired 2,6-isomer. However, it is also important in this preparation not to allow a large excess of
lithium diisopropylamide to remain in the reaction mixture; this base reacts with
benzyl bromide, forming
trans-stilbene8 which is difficult to separate from the reaction product.
14.
During the early part of the distillation when a substantial amount of
benzyl bromide is present, serious discoloration can be avoided by not heating the still pot above 140°. When the bulk of the
benzyl bromide has been removed, the temperature of the still pot may be raised to 150–160° to facilitate distillation of the product.
15.
The proportions of the desired 2,6-isomer and the unwanted 2,2-isomer in the alkylated product will vary depending on the rate and efficiency of mixing of the
benzyl bromide with the lithium enolate. If the alkylation of the initially formed enolate could be effected without any enolate equilibration, less than 2% of the unwanted 2,2-isomer would be expected.
916.
Sodium methoxide was purchased from Matheson, Coleman and Bell. Material from a freshly opened bottle was used without further purification.
17.
Commercial ethyl formate (Eastman Organic Chemicals) was purified by stirring it successively over anhydrous
sodium carbonate and over anhydrous
magnesium sulfate. The material was distilled to separate pure
ethyl formate, b.p.
54–54.5°.
3. Discussion
2-Benzyl-6-methylcyclohexanone has been prepared by the hydrogenation of
2-benzylidene-6-methylcyclohexanone over a
platinum or
nickel catalyst,
10 and by the alkylation of the sodium enolate of
2-formyl-6-methylcyclohexanone with
benzyl iodide followed by cleavage of the formyl group with aqueous base.
11 The 2,6-isomer was also obtained as a minor product (about 10% of the monoalkylated product) along with the major product,
2-benzyl-2-methylcyclohexanone by successive treatment of
2-methylcyclohexanone with
sodium amide and then with
benzyl chloride or
benzyl bromide12,13. Reaction of the sodium enolate of
2-formyl-6-methylcyclohexanone with
potassium amide in
liquid ammonia formed the corresponding dianion which when first treated with 1 equiv. of
benzyl chloride, then deformylated with aqueous base gave
2-benzyl-2-methylcyclohexanone14.
These synthetic routes illustrate the classical methods which have been used for the alkylation of unsymmetrical ketones. Reaction of the ketone with a strong base such as
sodium amide under conditions which permit equilibration of the enolates affords an equilibrium mixture of enolates, and subsequent reaction with an alkylating agent yields a mixture of monoalkylated products, as well as polyalkylated products. In the present case, the equilibrium mixture of metal enolates from
2-methylcyclohexanone contains 10–35% of the less highly substituted double bond isomer.
2,15,16 Consequently, the major alkylation product from this mixture is the 2,2-isomer. If the methylene group is protected with a blocking group, the resulting ketone is alkylated solely at the more highly substituted alpha
carbon. Removal of the blocking group affords pure 2,2-isomer. Alternatively, an activating group such as a formyl group or a carboalkoxyl group can be introduced at the less highly substituted alpha
carbon to permit selective alkylation at this position; the activating group is then removed.
An alternative solution to the problem of effecting the selective alkylation of an unsymmetrical ketone consists of generating a specific enolate under conditions where the enolate isomers do not equilibrate.
17 The methods which have been used to generate specific enolate anions include the reduction of enones
18,19 or α-haloketones
20,21 with metals, the reaction of organolithium reagents with enol silyl ethers
9,22,23 or enol esters,
17,23 and the kinetically controlled abstraction of the least hindered alpha proton from a ketone with a hindered base such as
lithium diisopropylamide.
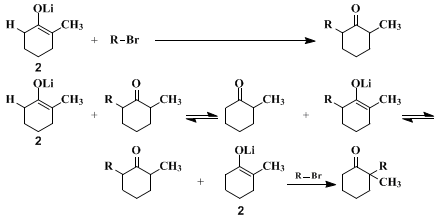
9 The present procedures illustrate the last two methods. To prevent the equilibration of lithium enolates during their formation, care is taken that no proton-donor BH (such as an alcohol or the un-ionized ketone) is present. Although with attention to this precaution, either of the structurally isomeric enolate ions can be prepared and maintained in solution, this fact does not ensure a structurally specific alkylation. As the accompanying equations illustrate, once reaction of the enolate with the alkyl halide is initiated, the reaction mixture will necessarily contain an un-ionized ketone, namely the alkylated product, and equilibration of the enolate ions can occur. Consequently, a structurally specific alkylation of an enolate anion can be successful only if the alkylation reaction is more rapid than equilibration so that the starting enolate
2 is consumed by the alkylating agent before significant amounts of the unwanted enolate
1 have been formed. In practice, this criterion normally is fulfilled with very reactive alkylating agents such as
methyl iodide. With less reactive alkylating agents such as
benzyl bromide and
n-alkyl iodides, some equilibration is usually observed.
23 The problem is aggravated when the alkylation involves the less stable and/or the less reactive enolate isomer (
e.g.,
2,). In the present procedures, relatively high concentrations of the enolate and the alkyl halide are employed to increase the alkylation rate and, consequently, decrease the proportion of the unwanted monoalkylation product which results from equilibration prior to alkylation. As is to be expected from the foregoing discussion, the alkylation of the enolate
1, forming
2-benzyl-2-methylcyclohexanone, exhibits more structural specificity than the alkylation of the enolate
2, forming
2-benzyl-6-methylcyclohexanone. The unwanted 2,2-isomer (
10–14%) in this alkylation is removed by a well-known chemical separation procedure in which the 2,2-isomer is converted to its formyl derivative.
24,25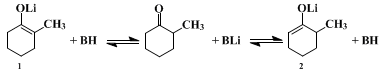
This preparation is referenced from:
- Org. Syn. Coll. Vol. 6, 248
- Org. Syn. Coll. Vol. 6, 478
- Org. Syn. Coll. Vol. 6, 571
- Org. Syn. Coll. Vol. 6, 666
- Org. Syn. Coll. Vol. 6, 692
- Org. Syn. Coll. Vol. 6, 762
- Org. Syn. Coll. Vol. 7, 172
- Org. Syn. Coll. Vol. 7, 346
- Org. Syn. Coll. Vol. 7, 351
- Org. Syn. Coll. Vol. 7, 386
- Org. Syn. Coll. Vol. 9, 328
- Org. Syn. Coll. Vol. 10, 12
- Org. Syn. Coll. Vol. 10, 59
Appendix
Chemical Abstracts Nomenclature (Collective Index Number);
(Registry Number)
methyllithium bipyridyl
hydrochloric acid (7647-01-0)
acetic acid (64-19-7)
ammonia (7664-41-7)
ether,
diethyl ether (60-29-7)
acetic anhydride (108-24-7)
sodium hydroxide (1310-73-2)
sodium hydrogen carbonate (144-55-8)
sodium carbonate (497-19-8)
oxygen (7782-44-7)
carbon tetrachloride (56-23-5)
nitrogen (7727-37-9)
platinum (7440-06-4)
nickel (7440-02-0)
sodium methoxide (124-41-4)
carbon (7782-42-5)
sodium (13966-32-0)
benzyl chloride (100-44-7)
methyl bromide (74-83-9)
ammonium hydroxide (1336-21-6)
p-xylene (106-42-3)
Methyl iodide (74-88-4)
ethyl formate (109-94-4)
Pentane (109-66-0)
magnesium sulfate (7487-88-9)
benzyl bromide (100-39-0)
sodium amide (7782-92-5)
lithium aluminum hydride (16853-85-3)
potassium amide
2-methylcyclohexanone (583-60-8)
bibenzyl (103-29-7)
Methyllithium (917-54-4)
perchloric acid (7601-90-3)
calcium hydride (7789-78-8)
1,2-dimethoxyethane (110-71-4)
2-Benzyl-2-methylcyclohexanone,
benzylmethylcyclohexanone,
Cyclohexanone, 2-methyl-2-(phenylmethyl)- (1206-21-9)
2-Butanol (78-92-2)
2,2'-bipyridyl (366-18-7)
lithium diisopropylamide (4111-54-0)
2-Benzyl-6-methylcyclohexanone,
cyclohexanone, 2-methyl-6-(phenylmethyl)- (24785-76-0)
2-Methyl-1-cyclohexen-1-yl acetate (75411-49-3)
diisopropylamine (108-18-9)
6-methyl-1-cyclohexen-1-yl acetate
2-benzylidene-6-methylcyclohexanone
2-formyl-6-methylcyclohexanone
benzyl iodide (620-05-3)
trans-Stilbene (103-30-0)
trans-2-benzyl-6-methylcyclohexanone
Copyright © 1921-, Organic Syntheses, Inc. All Rights Reserved