Org. Synth. 1973, 53, 86
DOI: 10.15227/orgsyn.053.0086
MODIFIED CLEMMENSEN REDUCTION: CHOLESTANE
Submitted by Shosuke Yamamura
1, Masaaki Toda
2, and Yoshimasa Hirata
2.
Checked by A. Laurenzano, L. A. Dolan, and A. Brossi.
1. Procedure
A
500-ml., four-necked, round-bottomed flask (Note 1) equipped with a sealed
mechanical stirrer (Note 2), a
gas-inlet tube, a
low-temperature thermometer, and a
calcium chloride tube is charged with
250 ml. of dry diethyl ether. With an
acetone–dry ice bath the temperature of the
ether is lowered to −10 to −15° and maintained within this range while a slow stream
(Note 3) of
hydrogen chloride is introduced, with slow stirring, for about 45 minutes. The gas-inlet tube is replaced with a
glass stopper, and
10.0 g. (0.0259 mole) of cholestan-3-one (Note 4) is added while the temperature of the stirred solution
(Note 5) is kept below −15°. The reaction mixture is cooled to −20°, and
12.3 g. (0.188 g.-atom) of activated zinc (Note 6) is added over a 2–3 minute period. The temperature of the reaction mixture is allowed to rise to −5°
(Note 7), and it is maintained between −4° and 0°
(Note 8) for 2 hours. Stirring is not interrupted for the duration of the reaction. The mixture is finally cooled to −15° and poured slowly onto about 130 g. of crushed ice. The ethereal layer is separated, and the aqueous layer is extracted with
100 ml. of ether that had been used to rinse the reaction vessel. The ethereal solutions are combined, washed with saturated aqueous
sodium chloride, dried over anhydrous
magnesium sulfate, and filtered. The ether is distilled under reduced pressure with a 50°
water bath, leaving
9.3–9.5 g. of a colorless, liquid residue that solidifies on cooling. This solid is dissolved in
30–40 ml. of n-hexane (Note 9). The solution is poured onto a
3.5 cm. by 17 cm. column of silica gel
(Note 10) and eluted with
80–90 ml. of n-hexane. Distillation of the solvent under reduced pressure with a 50° water bath leaves
8.0–8.2 g. (
82–84%) of
cholestane (Note 11), which, after recrystallization from
ethanol-ether (Note 12), yields
7.3–7.5 g. (
76–77%) of product as plates m.p.
78–79° (lit., m.p.
80°)
3 (Note 13).
2. Notes
1.
A standard,
three-necked flask fitted with a
Y-tube may be used.
2.
An
efficient magnetic stirrer may be substituted.
3.
Approximately one bubble per second can be spot-checked periodically by connecting the calcium chloride tube to an oil-filled bubble counter.
5.
The
cholestanone does not dissolve completely at this low temperature, but the reaction is not affected.
6.
The submitters prepared activated
zinc by the following procedure.
Commercial zinc powder (16 g.), special grade, ca. 300 mesh, obtained from either Kishida Chemical Company Ltd. or Hayashi Pure Chemical Company Ltd., is added with stirring to a
300-ml., round-bottomed flask containing
100 ml. of 2% hydrochloric acid. Vigorous stirring is continued until the surface of the
zinc becomes bright (
ca. 4 minutes). The aqueous solution is decanted, and the
zinc powder in the flask is washed by decantation with four 200-ml. portions of distilled water. The activated
zinc powder is transferred to a
suction filter with 200 ml. of distilled water and washed successively with
50 ml. of ethanol,
100 ml. of acetone, and
50 ml. of dry ether. Filtration and washing should be done as rapidly as possible to minimize exposure of the activated
zinc to air. The
zinc is finally dried at 85–90° for 10 minutes in a vacuum
oven (
ca. 15 mm.), cooled, and used immediately; the yield is 13–14 g.
The checkers used this procedure with certified zinc powder, 325 mesh, obtained from Fisher Scientific Company.
7.
This requires
ca. 20 minutes.
8.
The temperature is regulated by adding pieces of dry ice to the cooling bath as required. As the reduction proceeds, the solution separates into two phases.
9.
The solution is decanted from any insoluble matter.
10.
Silicic acid, 100 mesh (Mallinckrodt), was used.
11.
This material melts at
78–79°. On TLC [silica, development with
n-hexane, visualization with
sulfuric acid-methanol (1:1) and heating] the product had
Rf = 0.74. An impurity,
Rf = 0.65, was present.
12.
The
cholestane is dissolved in
50 ml. of ether.
Ether is distilled until the volume is 25 ml.,
200 ml. of ethanol is added, and the mixture is refrigerated.
13.
Recovery is
92%. Recrystallization has no effect on the quality of the product as judged by m.p. and TLC
(Note 11).
3. Discussion
The well-known Clemmensen reduction
4 is a general method by which aralkyl ketones are readily converted to the corresponding hydrocarbons with amalgamated
zinc and
hydrochloric acid. It is not particularly effective, however, with alicyclic and aliphatic ketones. The procedure described herein provides a simple method of reducing a variety of ketones to their desoxy derivatives in high yields under much milder conditions (0°, 1–2 hours) than those normally used in the Clemmensen reaction.
4 This permits selective deoxygenation of ketones in polyfunctional molecules
5 containing groups such as cyano, amido,
acetoxy, and carboalkoxy, which are stable under the mild reaction conditions. For example, the following reduction
6 has been carried out successfully by the modification of our procedure, using
acetic anhydride as the solvent.
5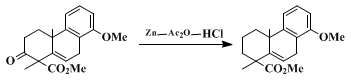
Wide latitude is permitted in choosing the solvent for the reaction. Several organic solvents (
tetrahydrofuran,
benzene,
n-hexane)
7 and particularly
acetic anhydride5,8 may be used instead of dry ether. α-Halo- and α-acetoxycholestanone
5 are converted to
cholestane with Zn-HCl-Et
2O and also with Zn-HCl-Ac
2O.
7,8 These reduction systems, however, have given different results with α,β-unsaturated ketones.
9 With Zn-HCl-Et
2O,
cholest-1-en-3-one gave
cholestane in
88% yield, while
cholest-4-en-3-one gave an 88% yield of a mixture of 1.2 parts of
cholestane and 1 part of
coprostane. By contrast, reaction of Zn-HCl-Ac
2O with
cholest-1-en-3-one afforded a mixture of three compounds:
cholestane (
30–32%),
3-acetoxycholest-2-ene (
10–24%), and
cholestan-3-one (
30–40%).
Cholestan-3-one appears to be formed from the corresponding
cyclopropanol acetate10 during the work up. The mechanism of this reduction is probably similar to that of the Clemmensen reaction.
11
Appendix
Chemical Abstracts Nomenclature (Collective Index Number);
(Registry Number)
ethanol (64-17-5)
hydrogen chloride,
hydrochloric acid (7647-01-0)
Benzene (71-43-2)
ether,
diethyl ether (60-29-7)
acetic anhydride (108-24-7)
sodium chloride (7647-14-5)
acetone (67-64-1)
zinc (7440-66-6)
magnesium sulfate (7487-88-9)
Cholestanone,
cholestan-3-one (566-88-1)
Tetrahydrofuran (109-99-9)
n-hexane (110-54-3)
cholest-1-en-3-one
Cholest-4-en-3-one (601-57-0)
acetoxy
Cholestane,
coprostane (481-21-0)
sulfuric acid-methanol
3-acetoxycholest-2-ene
cyclopropanol acetate
Copyright © 1921-, Organic Syntheses, Inc. All Rights Reserved