Org. Synth. 1976, 55, 127
DOI: 10.15227/orgsyn.055.0127
SULFIDE CONTRACTION via ALKYLATIVE COUPLING: 3-METHYL-2,4-HEPTANEDIONE
[2,4-Heptanedione, 3-methyl-]
Submitted by P. Loeliger
1 and E. Flückiger
2.
Checked by K. Matsuo and G. Büchi.
1. Procedure
Caution! To avoid exposure to toxic dichlorophenylphosphine vapors, the Grignard reaction should be conducted in a hood.
Benzene has been identified as a carcinogen; OSHA has issued emergency standards on its use. All procedures involving benzene should be carried out in a well-ventilated hood, and glove protection is required.
A. Bis(3-dimethylaminopropyl)phenylphosphine. A 3-l. separatory funnel is charged with 395 g. (2.50 moles) of 3-chloro-N,N-dimethyl-1-propylamine hydrochloride (Note 1), and a cold solution of 179 g. of potassium hydroxide in 540 ml. of water is added. The mixture is extracted three times with 300-ml portions of 5:1 diethyl ether-dichloromethane. The organic extracts are washed with 300 ml. of aqueous 2 N potassium hydroxide, combined, and dried over anhydrous sodium sulfate. The solvent is removed by distillation through a 25-cm. Vigreux column at atmospheric pressure, and the residual liquid is distilled under reduced pressure through a 13-cm. Vigreux column, giving 263–276 g. (87–91%) of 3-chloro-N,N-dimethyl-1-propylamine as a colorless liquid, b.p. 72–73° (100 mm.) (Note 2) and (Note 3), which is used immediately in the Grignard reaction (Note 4).
A 3-l., four-necked, round-bottomed flask equipped with a sealed mechanical stirrer, a pressure-equalizing dropping funnel, a thermometer, and a condenser fitted with a nitrogen-inlet tube is charged with 48.6 g. (2.00 g.-atoms) of magnesium turnings (Note 5). The flask is flushed with dry nitrogen and thoroughly dried with a heat gun, and 300 ml. of anhydrous tetrahydrofuran (Note 6) is added. The Grignard reaction is initiated by adding about 10% of a solution of 243.0 g. (1.98 moles) of 3-chloro-N,N-dimethyl-1-propylamine in 300 ml. of anhydrous tetrahydrofuran (Note 6), and 4 ml. of ethyl bromide while gently heating the flask with the drier (Note 7). The remainder of the 3-chloro-N,N-dimethyl-1-propylamine solution is added over a period of approximately 1 hour so as to maintain gentle reflux. The reaction mixture is heated at reflux for 3 hours, after which time most of the magnesium has reacted. The dark gray solution is cooled to 0° before a solution of 107.3 g. (81.29 ml., 0.5994 mole) of dichlorophenylphosphine (Note 8) in 200 ml. of anhydrous tetrahydrofuran (Note 6) is added dropwise, with efficient stirring, over a 1 hour period so that the temperature does not exceed 5° (Note 9). A greenish precipitate is formed locally where the phosphine is added. After the addition is complete, the reaction mixture is stirred and heated at reflux for 2 hours, during which time a heavy, greenish precipitate is formed. After cooling to room temperature, 600 ml. of ether (Note 10) is added, and the reaction mixture is left standing overnight, during which time the precipitate separates to the bottom of the flask. The solution is decanted into a 3-l. separatory funnel containing 300 ml. of 40% aqueous potassium hydroxide and 1 kg. of ice. The remainder of the reaction product is suction filtered with the aid of 1200 ml. of 5:1 ether–dichloromethane through a 3-cm. layer of Celite® (Note 11). The filtrate is added to the separatory funnel, and the organic layer is separated and washed twice with 600-ml. portions of saturated aqueous sodium chloride. The aqueous layer is extracted four times with 700-ml. portions of 5:1 ether–dichloromethane. The combined organic extracts are dried over anhydrous sodium sulfate, and the solvent is removed with a rotary evaporator. The crude yellow oil is distilled at high vacuum through a 14-cm. Vigreux column, yielding 109–116 g. (65–69%, based on phenylphosphonous dichloride) of bis(3-dimethylaminopropyl)phenylphosphine as a colorless liquid, b.p. 100–108° (0.005 mm.) (Note 12). Redistillation furnishes 94–97 g. (56–58%) of product, b.p. 102–105° (0.005 mm.) (Note 13), n24D 1.5265.
B. S-(2-Oxobut-3-yl) Butanethioate. A 750-ml., four-necked, round-bottomed flask equipped with a sealed mechanical stirrer, a pressure-equalizing dropping funnel, a thermometer, and a condenser fitted with a nitrogen-inlet tube is charged with 10.4 g. (0.100 mole) of thiobutyric acid (Note 14) in 300 ml. of anhydrous ether (Note 10). With stirring, 10.1 g. (0.100 mole) of triethylamine (Note 15) is added in one portion. Over a 15-minute period (Note 16), 15.1 g. (0.100 mole) of 3-bromo-2-butanone (Note 17) is added dropwise from the dropping funnel. The solution is heated at reflux with stirring for 1.5 hours and filtered through Celite®; the precipitate is washed with 60 ml. of ether. The ether solution is concentrated on a rotary evaporator. The residual orange-yellow oil is dissolved in 20 ml. of 5:1 benzene–ether and filtered through 70 g. of silica gel (Note 18), using 500 ml. of this solvent mixture as eluent. The solvent is removed on a rotary evaporator, yielding 17.0–17.4 g. (98–100%) of the thiol ester as a pale yellow oil which can be used without further purification in the next step. (Note 19) and (Note 20).
C. 3-Methyl-2,4-heptanedione. A dry, 500-ml., three-necked, round-bottomed flask equipped with a magnetic stirring bar, a pressure-equalizing dropping funnel, a thermometer, and a condenser fitted with a nitrogen-inlet tube is charged with 17.8 g. (0.204 mole) of anhydrous lithium bromide (Note 21). Under a nitrogen atmosphere, 34.8 g. (0.218 mole) of S-(2-oxobut-3-yl)butanethioate dissolved in 120 ml. of anhydrous acetonitrile (Note 22) is added to the flask. With stirring, the mixture is heated with a drier until a homogeneous solution is obtained. From the dropping funnel, 67 g. (69 ml., 0.24 mole) of redistilled bis(3-dimethylaminopropyl)phenylphosphine is added in one portion to the warm (ca. 60°) solution. The temperature rises to about 70°, and after 1–2 minutes a thick, white precipitate appears. The reaction mixture is stirred at 70° for 15 hours (Note 23). After cooling to room temperature, the reaction mixture is transferred with 600 ml. of 5:1 ether–dichloromethane into a separatory funnel containing 900 ml. of cold 1 N hydrochloric acid. The organic layer is separated and washed three times with 500-ml. portions of saturated aqueous sodium chloride. The aqueous phase is washed twice with 600-ml. portions of 5:1 ether–dichloromethane. The combined organic layer is dried over anhydrous sodium sulfate, and the solvent removed on a rotary evaporator, the temperature of the bath not exceeding 30°. The crude yellow oil is distilled through a 10-cm. Vigreux column under reduced pressure, yielding 23.5–24.7 g. (83–87%) of 3-methyl-2,4-heptanedione as a colorless liquid, b.p. 74–76° (9 mm.), n23D 1.4455 (Note 24).
2. Notes
1.
3-Chloro-N,N-dimethyl-1-propylamine hydrochloride was purchased from Fluka AG CH-9470 Buchs or Aldrich Chemical Company, Inc.2.
The fractions may be analyzed by GC for absence of solvent; a
300 cm. by 0.3 cm. glass column packed with XE-60 (1.5% w/w) coated on Chromosorb G AW DCMS (80/100 mesh) was employed.
3.
The spectral properties of the product are as follows; IR (neat) cm.
−1: 1470, 1465, 1265, 1040;
1H NMR (CDCl
3), δ (multiplicity, coupling constant
J in Hz., number of protons): 1.8–2.6 (m, 4H), 2.2 (s, 6H), 3.6 (t,
J = 6, 2H).
4.
It is advisable to distill the solvent one day, store the residue overnight under
nitrogen at 0°, and distill the product the next morning, allowing ample time for the following Grignard reaction. On standing at room temperature a white solid precipitates.
5.
Magnesium turnings were purchased from E. Merck & Company, Inc., Darmstadt, Germany or J. T. Baker Chemical Company.
6.
Tetrahydrofuran (purchased from Fluka AG or J. T. Baker Chemical Company) was distilled from
sodium hydride prior to use. For warnings regarding the purification of
tetrahydrofuran, see
Org. Synth., Coll. Vol. 5, 976 (1973).
7.
It is advisable to have available an
ice bath for cooling, should the reaction become violent.
8.
Dichlorophenylphosphine was purchased from Fluka AG or Aldrich Chemical Company, Inc.9.
The reaction is very exothermic and cooling with an
ice–sodium chloride bath is necessary.
10.
Anhydrous
ether was purchased from Fluka AG or J. T. Baker Chemical Company.
11.
During this operation, the reaction vessel is washed with 5:1
ether–
dichloromethane several times.
12.
The colorless forerun weighs
12–20 g.; the dark brown residue weighs
13–22 g.13.
The reported b.p. is
102–105° (0.005 mm.); a full spectroscopic characterization is given in the original paper.
314.
The
thiobutyric acid was prepared
4 as follows: A rapid stream of
hydrogen sulfide is passed, with vigorous stirring at −30°, through
200 ml. of anhydrous pyridine, contained in a
four-necked, round-bottomed flask equipped with a sealed mechanical stirrer, a
gas-inlet tube, a pressure-equalizing dropping funnel, and a thermometer. Over approximately 1 hour,
50 g. of 1-butyryl chloride is added dropwise to this solution. Approximately
400 ml. of 5 N sulfuric acid is added slowly until the pH is ~5. The organic acid, which separates as a yellow oil, is taken up in
ether and dried over anhydrous
sodium sulfate. After removal of the ether on a rotary evaporator, the product is distilled through a 14-cm. Vigreux column under a
nitrogen atmosphere, yielding
23.1–32.1 g. (
43–65%, not optimized by the submitters) of
thiobutyric acid as a colorless liquid, b.p.
119–121°.
15.
Triethylamine was purchased from Fluka AG or J. T. Baker Chemical Company.
16.
A colorless precipitate of
triethylamine hydrobromide is formed immediately. The temperature rises to about 35°.
17.
3-Bromo-2-butanone was purchased from Fisher Scientific Company. The submitters prepared it according to the literature
5 and checked its purity (>95%) by GC
(Note 2).
18.
Silica gel (70–230 mesh ASTM) purchased from E. Merck & Company, Inc., Darmstadt, Germany was used in a 2.5-cm. diameter column.
19.
GC analysis
(Note 2) indicated <2% impurities. IR (neat) cm.
−1: 1720, 1695.
20.
The submitters obtained a similar yield on ten times the scale.
21.
The absence of water in the
lithium bromide is of great importance. Traces of water lower the yield of product by 10–20%.
Lithium bromide dihydride (purchased from E. Merck & Company, Inc., Darmstadt or City Chemical Corporation) was dissolved three times in anhydrous 1:1
acetonitrile–
benzene, and the solvents were removed each time with a rotary evaporator. The
lithium bromide was dried under high vacuum at 100° for 1 hour, ground to a fine powder with a
mortar and pestle while still warm, and again dried at 100°, as above, for 3 hours.
22.
Acetonitrile was purchased from Fluka AG or J. T. Baker Chemical Company and distilled from
potassium carbonate immediately prior to use.
23.
The reaction is followed best by GC analysis
(Note 2). Traces of water seem to slow down the rate of the reaction.
24.
By GC analysis
(Note 2) the product is >98% pure. In the literature,
3 a full spectroscopic characterization is given. IR (neat) cm.
−1: 1725, 1700, 1600, 1360.
3. Discussion
This procedure illustrates a broadly applicable method which is essentially that found in the literature
3 for the synthesis of enolizable β-dicarbonyl compounds.
3 Although there are various methods for the preparation of β-dicarbonyl systems,
6 sulfide contraction widens the spectrum of available methods. The procedure can also be utilized in the sycnthesis of aza and diaza analogs of β-dicarbonyl systems. Eschenmoser
3 has utilized the method to produce vinylogous amides and amidines in connection with the total synthesis of corrins and vitamin B
12.
7,8S-Alkylation of a thiocarboxylic acid with an α-halogenated carbonyl compound gives a thiol ester in which the two carbons to be connected are linked
via a sulfur bridge (see the scheme below). Enolization and formation of the episulfide creates the desired carbon–carbon bond. Removal of atomic sulfur by a thiophile, either a phosphine or a phosphite, liberates the β-dicarbonyl compound. The addition of base is necessary in most cases; however, in the vinylogous amidine systems
7,8 electrophilic catalysis was employed. Normally a teritary alkoxide is utilized in the contraction. The addition of anhydrous
lithium bromide or
lithium perchlorate allows the reaction to proceed with the use of a tertiary amine as the base. Presumably, the lithium salts complex with the carbonyl groups, enhancing the enolization and/or contraction step.
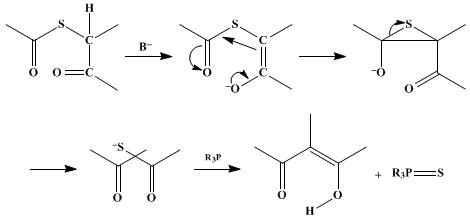
This procedure also incorporates the use of bis(3-dimethylaminopropyl)phenylphosphine as a combined amine–phosphine reagent. The merit of using this basic phosphine as opposed to a tertiary amine and a phosphine lies in the ease of workup. Excess phosphine and phosphine sulfide can be removed by extraction with dilute acid.
Since the new carbon–carbon bond is formed intramolecularly in the sulfide extrusion method, its main potential lies in cases where intermolecular condensations fail.
9,10
Appendix
Chemical Abstracts Nomenclature (Collective Index Number);
(Registry Number)
Celite
diethyl ether-dichloromethane
potassium carbonate (584-08-7)
sulfuric acid (7664-93-9)
hydrochloric acid (7647-01-0)
Benzene (71-43-2)
ether (60-29-7)
acetonitrile (75-05-8)
magnesium,
magnesium turnings (7439-95-4)
sodium chloride (7647-14-5)
hydrogen sulfide (7783-06-4)
Ethyl bromide (74-96-4)
sodium sulfate (7757-82-6)
nitrogen (7727-37-9)
pyridine (110-86-1)
potassium hydroxide (1310-58-3)
1-butyryl chloride (141-75-3)
dichloromethane (75-09-2)
Tetrahydrofuran (109-99-9)
sodium hydride (7646-69-7)
phosphine (7723-14-0)
triethylamine (121-44-8)
phenylphosphonous dichloride,
dichlorophenylphosphine (644-97-3)
triethylamine hydrobromide (636-70-4)
lithium bromide (7550-35-8)
3-Methyl-2,4-heptanedione,
2,4-Heptanedione, 3-methyl- (13152-54-0)
Bis(3-dimethylaminopropyl)phenylphosphine (32357-32-7)
thiobutyric acid (3931-64-4)
3-bromo-2-butanone (814-75-5)
Lithium bromide dihydride
lithium perchlorate (7791-03-9)
3-chloro-N,N-dimethyl-1-propylamine hydrochloride (5407-04-5)
3-chloro-N,N-dimethyl-1-propylamine
S-(2-oxobut-3-yl)butanethioate,
S-(2-Oxobut-3-yl) butanethioate (32425-98-2)
Copyright © 1921-, Organic Syntheses, Inc. All Rights Reserved