Org. Synth. 1979, 59, 102
DOI: 10.15227/orgsyn.059.0102
NUCLEOPHILIC ACYLATION WITH DISODIUM TETRACARBONYLFERRATE: METHYL 7-OXOHEPTANOATE AND METHYL 7-OXOÖCTANOATE
[Octanoic acid, 7-oxo-, methyl ester]
Submitted by Richard G. Finke
1,2
and Thomas N. Sorrell
13.
Checked by Richard T. Taylor and Martin F. Semmelhack.
1. Procedure
Caution! Iron pentacarbonyl and carbon monoxide are highly toxic; consequently all parts of this procedure should be carried out in a well-ventilated hood. Iron pentacarbonyl is easily recognized by its musty odor. Since disodium tetracarbonylferrate is very pyrophoric, the reagent must be kept under a dry inert atmosphere at all times.
Methyl iodide, in high concentrations for short periods and in low concentrations for long periods, can cause serious toxic effects in the central nervous system. Accordingly, the American Conference of Governmental Industrial Hygienists
4 has set 5 p.p.m., a level which cannot be detected by smell, as the highest average concentration in air to which workers should be exposed for long periods. The preparation and use of
methyl iodide should always be performed in a well-ventilated fume hood. Since the liquid can be absorbed through the skin, care should be taken to prevent contact.
A.
Disodium tetracarbonylferrate sesquidioxanate (Note 1). A
dry, 2-l., three-necked, round-bottomed flask is equipped with a mechanical stirrer (Note 2), a
three-way stopcock with one branch connected to a nitrogen source, and a Y-shaped adapter fitted with a reflux condenser vented through an oil bubbler and a pressure-equalizing dropping funnel capped by a rubber septum. The apparatus is flushed with
nitrogen (Note 3) for 15 minutes and charged with
600 ml. of dry, deoxygenated dioxane (Note 4) and
(Note 5),
10.6 g. (0.461 g.-atoms) of sodium, and
9.1 g. (0.050 mole) of benzophenone. The solution is stirred vigorously and heated under reflux with a heating mantle until the deep blue color of the benzophenone ketyl appears. With a
gas-tight syringe,
45.3 g. (29.8 ml., 0.231 mole) of iron pentacarbonyl (Note 6) is injected into the dropping funnel. The blue solution is then titrated to a white or slightly yellow end point by adding
iron pentacarbonyl to the refluxing solution over 2.5 hours
(Note 7). The suspension is heated at reflux for another 45 minutes, then cooled to room temperature. Precipitation of the
disodium tetracarbonylferrate sesquidioxanate as a white powder is completed by adding
600 ml. of dry, deoxygenated hexane (Note 5) and
(Note 8). The Y-shaped adapter and the mechanical stirrer are removed under a rapid stream of
nitrogen and quickly replaced with a
tight-fitting rubber septum and a
gas-dispersion tube with a fritted-glass tip (see
Figure 1). The solvent is forced up the gas-dispersion tube and out of the flask with
nitrogen pressure
(Note 9). The product is washed in the flask with two
400-ml. portions of hexane added
via a cannula
(Note 5), and the supernatant solvent is removed with
nitrogen pressure in the same manner. The
disodium tetracarbonylferrate sesquidioxanate is used directly in Parts B and C without drying or weighing
(Note 10).
B. Methyl 7-oxoheptanoate. In the same three-necked, round-bottomed flask containing 72–78 g. (0.21–0.23 mole) of disodium tetracarbonylferrate sesquidioxanate, the gas-dispersion tube is replaced with a mechanical stirrer, and 1.5 l. of dry, deoxygenated tetrahydrofuran (Note 11) is added. The light tan suspension is stirred vigorously as 41.8 g. (0.200 mole) of methyl 6-bromohexanoate (Note 12) is added in one portion by syringe. The nitrogen is flushed from the flask with carbon monoxide (Note 13) and (Note 14) admitted through the other branch of the three-way stopcock, and the suspension is stirred under 10 p.s.i. of carbon monoxide for at least 14 hours, during which time the solid dissolves. A rapid flow of nitrogen is swept through the flask while the septum is removed and replaced quickly with a pressure-equalizing dropping funnel. The dropping funnel is flushed with nitrogen and charged with 50 ml. of glacial acetic acid, added dropwise to the orange solution (Note 15). Stirring is continued for 20 minutes, after which the deep red solution is concentrated to a volume of ca. 400 ml. with a rotary evaporator in the hood (Caution! Some iron pentacarbonyl is present) and poured into 2 l. of water. The mixture is extracted with four 400-ml. portions of diethyl ether, and the combined organic solutions are washed with 400 ml. of water. The ethereal solution is mixed with 400 ml. of 2 M hydrochloric acid, and 68 g. of iron(III) chloride is added in small portions until carbon monoxide evolution subsides and the organic layer becomes green from triiron dodecacarbonyl. The organic layer is washed with successive 400-ml. portions of 2 M hydrochloric acid, water, saturated sodium hydrogen carbonate, and saturated sodium chloride. After being dried over anhydrous sodium sulfate, the ethereal solution is concentrated to a green oil with a rotary evaporator. Iron-containing by-products such as triiron dodecacarbonyl and iron pentacarbonyl are removed by rapid chromatography on a 7 × 40 cm. column prepared in hexane with 400 g. of silica gel (Note 16). The green triiron dodecacarbonyl is first eluted with ca. 3 l. of hexane before the product is eluted with 3 l. of 2:1 (v/v) ether–hexane. The ether–hexane eluate is dried over anhydrous magnesium sulfate and evaporated. Distillation of the residual oil through a 15-cm. Vigreux column at reduced pressure affords a small forerun of 1 ml. or less and 17.9–20.0 g. (57–63%) of methyl 7-oxoheptanoate, b.p. 65–80° (0.1 mm.), n20D 1.4388 (Note 17).
C. Methyl 7-oxoöctanoate. The 2-l., three-necked, round-bottomed flask containing 72–78 g. (0.21–0.23 mole) of disodium tetracarbonylferrate from Part A is flushed rapidly with nitrogen while the gas-dispersion tube is removed, a magnetic stirring bar is placed inside, and a pressure-equalizing dropping funnel is attached. The other branch of the three-way stopcock is connected to a bubbler, the dropping funnel is capped with a rubber septum, and 600 ml. of dry, deoxygenated N-methylpyrrolidinone (Note 5) and (Note 18) is added. The suspension is stirred and maintained under a static nitrogen atmosphere as 41.8 g. (0.200 mole) of methyl 6-bromohexanoate (Note 12) is injected into the dropping funnel with a gas-tight syringe and added dropwise into the flask. The resulting solution (Note 19) is stirred for 30 minutes at room temperature and cooled in an ice bath before 64 g. (28 ml., 0.45 mole) of methyl iodide is added over 20 minutes. The ice bath is removed, and stirring is continued for 20–40 hours (Note 14). The dark-red mixture is poured into 3 l. of saturated aqueous sodium chloride and extracted with three 400-ml. portions of ether and one 400-ml. portion of hexane. The combined organic solutions are washed with one 400-ml. portion of water and one 400-ml. portion of saturated sodium chloride. After 400 ml. of 2 M hydrochloric acid is added, the iron by-products are oxidized with iron(III) chloride exactly as described in Part B. The ethereal solution is evaporated under reduced pressure, the residual oil is applied to a column of silica gel packed in hexane, and the organic product is separated from the iron by-products by chromatography as described in Part B. The ether–hexane eluate is dried over anhydrous magnesium sulfate, the solvent is evaporated, and the remaining liquid is distilled at reduced pressure, giving 24.0–24.8 g. (70–72%) of methyl 7-oxoöctanoate, b.p. 112–127° (10 mm.), n20D 1.4360 (Note 20).
2. Notes
1.
Approximately 4.5 hours are required to complete Part A.
2.
A
mechanical stirrer is necessary on any scale owing to the formation of a thick slurry toward the end of the reaction.
3.
The submitters passed the
nitrogen through
ca. 100 g. of BASF catalyst R3-11 contained in a metal tube and heated at 160°, to remove
oxygen, and through a column of Linde type 3A molecular sieves, to remove water. The catalyst (catalog number 18-3000-00) and pertinent literature were obtained from Chemical Dynamics Corporation, P.O. Box 395, South Plainfield, New Jersey 07080. The checkers used “prepurified”
nitrogen and
argon in separate runs without additional drying or
oxygen scavenging.
4.
Dioxane was heated at reflux with
sodium overnight under
nitrogen,
benzophenone was added, and the solvent was distilled after appearance of the deep-blue color of the benzophenone ketyl.
5.
The submitters recommend that the solvent be distilled under
nitrogen into a
two-necked receiving flask fitted with a three-way stopcock. The receiving flask is separated from the distillation apparatus under a rapid
nitrogen flow and fitted quickly with a rubber septum. The solvent is then transferred to the reaction vessel by needlestock techniques
5 as follows: A stainless-steel cannula with a 2-mm. inside diameter and both ends sharpened is inserted through the septum into the receiving flask above the surface of the liquid, and a stream of
nitrogen is passed briefly through the stopcock and out the cannula, removing air. The other end of the cannula is then inserted through the septum on the reaction vessel, the end of the cannula in the receiver is pushed below the surface of the liquid, and the solvent is forced into the reaction vessel with
nitrogen pressure.
6.
Iron pentacarbonyl was purchased from PCR, Inc., Gainesville, Florida, and stored under
nitrogen.
7.
Approximately
95% of the iron pentacarbonyl is added within 2 hours, and the remaining 5% is then added dropwise over the next 30 minutes. The blue color should never be completely discharged prior to the end point, particularly toward the end of the reaction, since the remaining solution may be deactivated. Avoiding premature discharge of the blue color is especially important in small-scale preparations. At the end point
1 ml. or less of the iron pentacarbonyl remains in the dropping funnel. The checkers carried out the reaction on a smaller scale in two runs, adding
13.1 g. (8.62 ml., 0.0669 mole) of iron pentacarbonyl as a solution in
50 ml. of dry dioxane.
8.
Reagent grade hexane was dried and deoxygenated by distillation from
calcium hydride under
nitrogen.
9.
Alternatively,
300–400 ml. of the dioxane may be transferred by this method into another flask before the
hexane is added. The recovered
dioxane may then be used in another preparation without purification.
10.
The yield is typically
72–78 g. (
90–100%). An excess of the reagent is not detrimental to the procedures in Parts B and C. The submitters have doubled the scale of this procedure with no change in the yield. For smaller-scale reactions the submitters recommend that the reagent be purchased from Alfa Division, Ventron Corporation. The checkers used the commercially available reagent successfully in one run, the material having been transferred to the reaction vessel in a dry box.
11.
Tetrahydrofuran was dried and deoxygenated by distillation from
calcium hydride under
nitrogen.
12.
6-Bromohexanoic acid was purchased from Aldrich Chemical Company, Inc., and esterified with
sulfuric acid and
methanol.
Methyl 6-bromohexanoate was obtained as a colorless liquid, b.p.
92–94° (5 mm.),
n20D 1.4510, judged to be greater than 99% pure according to GC analysis on dimethylsilicone (OV-101) as liquid phase.
13.
A cylinder of
carbon monoxide equipped with a suitable regulator calibrated in pounds per square inch (p.s.i.) is connected to the three-way stopcock. All joints and the septum must be secured with clamps or wire. A vertical tube containing
mercury was connected to an exit tube from the reaction flask by the checkers. A pressure of
carbon monoxide was maintained against a
500-mm. column of mercury.
14.
Alkyl tetracarbonyl iron(0) reagents in solution decompose more rapidly with increasing concentration and temperature, especially above 0°.
Carbon monoxide must be added without delay to convert this intermediate to the more stable acyl iron compound.
15.
Subsequent operations may be conducted in air. However, the procedure should not be interrupted until the ethereal solution is drying over
sodium sulfate.
16.
Silica gel of mesh 60–200 was supplied by Davison Chemical Division, W. R. Grace and Company, Baltimore, Maryland, and dried at 70° before use. The flow rate of
hexane during the chromatography was 4 l. per hour.
17.
The checkers obtained
13.6 g. (
43%) of product, b.p.
65–75° (0.1 mm.). The submitters determined the purity of the product to be greater than 90% by GC using
10% dimethylsilicone (OV-101) as liquid phase at 200°. The
1H NMR spectrum (CDCl
3) shows absorptions at δ (multiplicity, coupling constant
J in Hz., number of protons, assignment): 1.48 (m, 6H, 3C
H2), 2.32 (t, 2H, C
H2CO
2CH
3), 2.40 (t of d,
J = 2 and 8, 2H, C
H2CH=O), 3.65 (s, 3H, CO
2C
H3), 9.62 (t,
J = 2, 1H, C
H=O).
18.
N-Methylpyrrolidinone was heated at reflux over
calcium hydride under reduced pressure for at least 24 hours by the submitters, then distilled from the
calcium hydride under reduced pressure. The checkers stirred the solvent in the presence of
calcium hydride at 100° for 48 hours prior to distillation at 150 mm.
19.
The homogeneous solution, if free of impurities, is bright yellow. Usually, however, the color is dark red or orange, evidently owing to the presence of trace amounts of impurities.
20.
Starting with
13.1 g. (8.62 ml., 0.0669 mole) of iron pentacarbonyl, the checkers obtained
7.3–7.4 g. (
71–75%) of product, b.p.
92–95° (2 mm.) A GC analysis by the submitters using
dimethylsilicone (OV-101) as liquid phase at 200° showed the purity of the product to be greater than 95%. The spectral properties of the product are as follows: IR (thin film) cm.
−1: 1735 (ester C=O), 1715 (ketone C=O);
1H NMR (CDCl
3), δ (multiplicity, coupling constant
J in Hz., number of protons, assignment): 1.46 (m, 6H, 3C
H2), 2.12 (s, 3H, CH
2COC
H3), 2.32 (t,
J = 8, 2H, C
H2CO
2CH
3), 2.44 (t,
J = 8, 2H, C
H2COCH
3), 3.67 (s, 3H, CO
2C
H3).
3. Discussion
This procedure describes the preparation of
disodium tetracarbonylferrate sesquidioxanate by reduction of
iron pentacarbonyl with
sodium and the use of the reagent for nucleophilic formylation and acylation of the primary bromide,
methyl 6-bromohexanoate.
Disodium tetracarbonylferrate serves as a synthetic equivalent of
carbon monoxide dianion,
, in most preparations. Thus, the reagent reacts with alkyl halides and
p-toluenesulfonates, forming anionic alkyltetracarbonyl iron(0) complexes which combine in a second step with various electrophiles, giving carbonyl compounds.
6 The reagent donates the new carbonyl carbon, which becomes bonded to both the alkyl group and the electrophile in the final product. If the electrophile is a proton from
acetic acid or an alkyl group from a second alkyl halide, the overall transformations amount to nucleophilic formylation
6 and acylation,
7 respectively. The preparation of carboxylic acids, esters, and amides by formal nucleophilic carboxylation is accomplished by use of
oxygen and water (or
sodium hypochlorite and water),
iodine and alcohols, and
iodine and amines, respectively, as the electrophiles.
8 The general reactions are summarized by the following equations and several specific examples
7,8,9 are presented in Table I.
TABLE I
NUCLEOPHILIC ACYLATION AND CARBOXYLATION OF ALKYL HALIDES AND p-TOLUENESULFONATES WITH DISODIUM TETRACARBONYLFERRATE7,8,9
|
|
Alkyl Halide or p-Toluenesulfonatea
|
Electrophile
|
Product
|
Yield (%)
|
|
|
C6H5(CH2)2Br
|
CH3CO2H
|
|
86b
|
|
(CH3)2C=CH(CH2)2Br
|
CH3CO2H
|
|
81b
|
|
|
CH3CO2H
|
|
50b,c
|
|
CH3(CH2)7Br
|
CH3CH2I
|
|
80
|
|
|
CH3I
|
|
79
|
|
CH3(CH2)5Br
|
O2,H2O
|
|
82
|
|
Cl(CH2)6Br
|
O2, H2O
|
|
84
|
|
CH3(CH2)7Br
|
I2, C2H5OH
|
|
84
|
|
CH3(CH2)4Br
|
I2, (C2H5)2NH
|
|
80b
|
|
aThe structural abbreviation Ts is used for p-toluenesulfonate.
|
|
|
cIsomeric octenes identified as by-products.
|
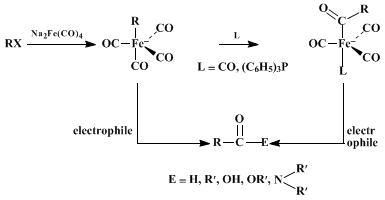
The initial reaction between the alkyl halide (or
p-toluenesulfonate) and
disodium tetracarbonylferrate behaves as a typical
SN2-type substitution.
6,10 Thus, this step proceeds smoothly with primary and secondary reactants, but the tertiary analogs fail owing to elimination. Allylic substrates are also incompatible, since these undergo elimination, forming stable iron tricarbonyl-1,3-diene complexes. The initial substitution with secondary
p-toluenesulfonates occurs more efficiently than with the corresponding halides, and the stereochemistry results from clean inversion. The solvents used are generally either
tetrahydrofuran or
N-methylpyrrolidinone, the reactions being as much as 10
4 times faster in the latter. The initially formed alkyl iron intermediate rearranges to an acyl iron complex either prior to or during the subsequent reaction with the electrophile.
6,7,8,9,11 The presence of
carbon monoxide or
triphenylphosphine enhances the rate of rearrangement to the more stable acyl iron intermediate. The reactions show high selectivity and are compatible with functional groups such as chloro, cyano, and esters. The reagent reacts with acid chlorides, forming the acyl iron complexes directly, which may then be hydrolyzed to aldehydes or utilized for nucleophilic acylation.
Methyl 7-oxoheptanoate has previously been synthesized from
cycloheptanone in two steps with a
42% overall yield.
12 7-Oxoöctanoic acid, the methyl ester of which is the product of Part C, has been prepared by base-induced ring cleavage of
2-acetylcyclohexanone.
13
Appendix
Chemical Abstracts Nomenclature (Collective Index Number);
(Registry Number)
silica gel
H2O
Disodium tetracarbonylferrate sesquidioxanate
DISODIUM TETRACARBONYLFERRATE
triiron dodecacarbonyl
dimethylsilicone
sulfuric acid (7664-93-9)
hydrochloric acid (7647-01-0)
acetic acid (64-19-7)
methanol (67-56-1)
ether,
diethyl ether (60-29-7)
carbon monoxide (630-08-0)
sodium hydrogen carbonate (144-55-8)
sodium chloride (7647-14-5)
sodium sulfate (7757-82-6)
oxygen (7782-44-7)
nitrogen (7727-37-9)
6-Bromohexanoic acid (4224-70-8)
mercury (7439-97-6)
iodine (7553-56-2)
Benzophenone (119-61-9)
sodium (13966-32-0)
iron(III) chloride (7705-08-0)
Methyl iodide (74-88-4)
sodium hypochlorite (7681-52-9)
magnesium sulfate (7487-88-9)
dioxane (123-91-1)
Tetrahydrofuran (109-99-9)
hexane (110-54-3)
Cycloheptanone (502-42-1)
argon (7440-37-1)
calcium hydride (7789-78-8)
iron pentacarbonyl
triphenylphosphine (603-35-0)
N-methylpyrrolidinone (872-50-4)
2-acetylcyclohexanone (874-23-7)
Methyl 7-oxoheptanoate (35376-00-2)
Octanoic acid, 7-oxo-, methyl ester,
Methyl 7-oxooctanoate (16493-42-8)
methyl 6-bromohexanoate
p-toluenesulfonate
Copyright © 1921-, Organic Syntheses, Inc. All Rights Reserved