Org. Synth. 1983, 61, 141
DOI: 10.15227/orgsyn.061.0141
GENERATION AND REACTIONS OF ALKENYLLITHIUM REAGENTS: 2-BUTYLBORNENE
Submitted by A. Richard Chamberlin, Ellen L. Liotta, and F. Thomas Bond
1.
Checked by Hiroko Masamune and Robert V. Stevens.
1. Procedure
A. d-Camphor 2,4,6-triisopropylbenzenesulfonylhydrazone. In a 500-mL Erlen-meyer flask equipped with a magnetic stirring bar is placed 66.0 g (0.22 mol) of 2,4,6-triisopropylbenzenesulfonylhydrazide (Note 1), 30.4 g (0.20 mol) of d-camphor (Note 2), 100 mL of freshly distilled acetonitrile, and 20.0 mL (0.24 mol) of concentrated hydrochloric acid. The resulting solution is stirred overnight while a granular solid precipitates. The white crystals are cooled at −10°C for 4 hr and collected by suction filtration, dissolved in 175 mL of dichloromethane, filtered to remove a small amount of insoluble material, and concentrated under reduced pressure on a rotary evaporator to give 60.8–63.4 g (70–73%) of a white solid, mp 196–199°C (dec) (Note 3).
B. 2-Butylbornene. A 1-L, three-necked flask is equipped with a 250-mL addition funnel (sealed with a rubber septum), a mechanical stirrer, and a rubber septum. The system is vented (via a hypodermic needle inserted through the addition funnel septum) through a mineral oil bubbler, and the apparatus is flame-dried while it is flushed with prepurified nitrogen introduced through the septum of the flask. The flask is charged with 40.0 g (0.092 mol) of d-camphor 2,4,6-triisopropylbenzenesulfonylhydrazone, resealed, and again flushed with nitrogen. Hexane, 200 mL, (Note 4), and 200 mL of tetramethylethylenediamine (Note 5) are added, and the stirred solution, under an atmosphere of nitrogen, is cooled to approximately −55°C with an ethanol–water(2:1)/dry ice bath. Using a Luer-Lok syringe, 158 mL (0.20 mol) of 1.29 M sec-butyllithium (Note 6) is transferred to the addition funnel. The solution is stirred rapidly and the sec-butyllithium added over a period of 15–20 min. The resulting orange solution is stirred for 2 hr, and the cold bath removed. After 20 min the flask is immersed in an ice bath until nitrogen evolution ceases (approximately 10 min).
To this stirred solution of 2-lithiobornene is added, via syringe, 15.2 g (0.11 mol) of butyl bromide (Note 7) over a 1-min period. The ice bath is then removed, and the reaction mixture is stirred at room temperature overnight. The mixture is poured into 500 mL of water. The layers are separated and the aqueous layer extracted with two 100-mL portions of ether. The combined organic extracts are washed with five 200-mL portions of water, one 50-mL portion of 1 N hydrochloric acid, and two 200-mL portions of water. The solution is dried over anhydrous magnesium sulfate, filtered, and concentrated on a rotary evaporator at aspirator pressure and room temperature. Distillation of the residual yellow liquid through a 20-cm Vigreux column affords 8.9–9.4 g (50–53%) of product as a colorless liquid, bp 57–59°C (0.5 mm), nD25 1.4664, [α]D25 − 10.7° (c MeOH, 0.0747) (Note 8).
2. Notes
1.
The submitters used material prepared following a literature procedure.
22.
d-Camphor was purchased from Eastman Kodak Co.,
[α]D25 + 39.5°.
3.
The
1H NMR spectrum is as follows: δ: 0.61 (s, 3 H), 0.86 (s, 6 H), 1.26 (overlapping doublets,
J = 6.7, 18 H), 1.4–2.2 (m, 7 H), 2.90 (septuplet,
J = 7, 1 H), 4.20 (septuplet,
J = 7, 2 H), 7.15 (s, 2 H).
4.
MCB, Inc. reagent-grade hexane was distilled from
lithium aluminum hydride.
5.
This compound was purchased from Aldrich Chemical Company, Inc., and distilled from
lithium aluminum hydride.
6.
The
sec-butyllithium was purchased from Alfa Products, Morton Thiokol, Inc. and standardized by double titration or
diphenylacetic acid titration. Other alkyllithium bases such as
butyllithium and
methyllithium cannot be substituted for the stronger
sec-butyllithium since larger amounts of
bornylene are formed because of incomplete dianion formation. Careful attention must be paid to stoichiometry in this reaction; failure to do so also results in increasing the amount of
bornylene formed.
Even under ideal conditions the NMR of crude product shows 20–30% bornylene, which, however, is easily separated from the desired product during distillation as a "forerun" which sublimes into the vacuum pump trap.
7.
Analytical reagent material was purchased from Mallinckrodt, Inc., and distilled from
calcium hydride.
8.
The
1H NMR spectrum (CDCl
3) is as follows: δ 0.74 (s, 3 H), 0.76 (s, 3 H), 0.94 (s, 3 H), 0.7–1.0 (broad m, 7 H), 1.4 (m, 4 H), 1.9 (m, 2 H), 2.19 ("t",
J = 4, 1 H), 5.51 (m, 1 H).
3. Discussion
The sequence described here illustrates a general procedure for converting ketones into alkylated olefins:
It is a modification of the Shapiro olefin synthesis
3 that allows the alkenyl anion intermediate to be trapped with primary halides and other electrophiles. Use of triisopropylbenzenesulfonylhydrazones as the
vinyllithium precursor
4 is an improvement over previously
4 used toluenesulfonylhydrazones,
5,6 which can be employed in the sequence provided excess
sec-butyllithium (typically 4.5 equiv) and alkyl halide (3.0 equiv) are used. Methyl ketones (e.g.,
acetone,
acetophenone,
2-octanone) can also be used and can be converted into their dianions using 2.2 equiv of the weaker base,
butyllithium. The conditions described above, with the slight modifications noted, have been used for a variety of ketones as shown in Table I.
The submitters have found that the hexane–tetramethylethylenediamine solvent system described above, which is required for toluenesulfonylhydrazones, may be replaced with tetrahydrofuran when triisopropylbenzenesulfonylhydrazones are used, provided that the electrophilic reagent is added to the alkenyllithium species as soon as it is formed (as indicated by cessation of nitrogen evolution).
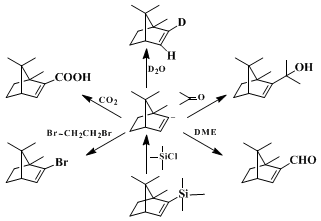
Primary alkyl bromides react well in this sequence except for particularly reactive compounds (e.g.,
methyl bromide,
allyl bromide) that give the vinyl halide by metal-halogen exchange. Secondary halides, as expected, suffer from elimination as a side reaction. Other electrophiles have been used successfully including D
2O, aldehydes and ketones,
dimethylformamide,
4,7 chlorotrimethylsilane,
4,8 1,2-dibromoethane,
4 and
carbon dioxide. Such sequences allow for relatively straightforward preparation of deuterated olefins, allylic alcohols, α,β-unsaturated aldehydes, alkenylsilanes, alkenyl bromides, and α,β-unsaturated acids. The major advantages of this route to alkenyllithium reagents
9 lie in the availability of the ketone precursors and the regiospecificity of the Shapiro reaction.
3,10 There are numerous alternative routes to trisubstituted olefins.
11
TABLE I
KETONE TO BUTYLALKENE CONVERSIONS
|
|
Ketone
|
Product
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
a sec-Butyllithium is added at −8°C.
|
b Approximately 2% of the isomeric 1-butyl-2-methylcyclohexene is formed.
|
c A mixture of (Z) and (E) isomers is formed.
|
d Tertiary hydrogen removal is slower. sec-Butyllithium (3.0 equiv) is added at −78°C; the solution is immediately warmed to room temperature and stirred for 1–2 hr before butyl bromide (2.0 equiv) is added.
|
Appendix
Chemical Abstracts Nomenclature (Collective Index Number);
(Registry Number)
M sec-butyllithium
hydrochloric acid (7647-01-0)
ether (60-29-7)
hydrogen (1333-74-0)
acetonitrile (75-05-8)
Allyl bromide (106-95-6)
Butyl bromide (109-65-9)
nitrogen (7727-37-9)
carbon dioxide (124-38-9)
acetone (67-64-1)
Acetophenone (98-86-2)
1,2-dibromoethane (106-93-4)
methyl bromide (74-83-9)
Diphenylacetic acid (117-34-0)
dichloromethane (75-09-2)
2-Octanone (111-13-7)
magnesium sulfate (7487-88-9)
d-camphor (21368-68-3)
butyllithium (109-72-8)
Tetrahydrofuran (109-99-9)
lithium aluminum hydride (16853-85-3)
dimethylformamide (68-12-2)
hexane (110-54-3)
Methyllithium (917-54-4)
calcium hydride (7789-78-8)
vinyllithium (917-57-7)
CHLOROTRIMETHYLSILANE (75-77-4)
tetramethylethylenediamine (20485-44-3)
sec-butyllithium (598-30-1)
2-Butylbornene (87068-33-5)
2,4,6-triisopropylbenzenesulfonylhydrazide (39085-59-1)
2-Lithiobornene
bornylene
1-butyl-2-methylcyclohexene
D-Camphor 2,4,6-triisopropylbenzenesulfonylhydrazone
Copyright © 1921-, Organic Syntheses, Inc. All Rights Reserved