Org. Synth. 1985, 63, 79
DOI: 10.15227/orgsyn.063.0079
2-METHYL-2-(TRIMETHYLSILOXY)PENTAN-3-ONE
[3-Pentanone, 2-methyl-2-[(trimethylsilyl)oxy]-]
Submitted by Steven D. Young, Charles T. Buse, and Clayton H. Heathcock
1.
Checked by Joseph R. Flisak, Sami Farahat, Stan S. Hall, Hugh W. Thompson, and Gabriel Saucy.
1. Procedure
A. 2-Hydroxybutanenitrile. A 3-L, three-necked, round-bottomed flask is fitted with a mechanical stirrer and thermometer and charged with 312 g (3.0 mol) of sodium bisulfite and 1050 mL of water. The stirrer is started and after the sodium bisulfite has dissolved, the flask is placed in an ice–salt bath. A solution of 147 g (3.0 mol) of sodium cyanide (Note 1) in 450 mL of water and 174 g (3.0 mol) of propanal (Note 2) are separately cooled to 0°C in ice–salt baths. When the temperature of the vigorously stirring sodium bisulfite solution has stabilized at 0°C the cold propanal is added in one portion. The temperature of the reaction solution immediately increases to ca. 35°C and then returns to ca. 0°C. After 30 min the cold sodium cyanide solution is added in one portion. The reaction mixture again warms to ca. 15°C and then returns to ca. 0°C. This mixture is stirred for 2 hr at 0°C, during which time a thick white precipitate of sodium sulfite forms. The supernatant liquid is decanted into a 4-L separatory funnel and the precipitate is washed with 1 L of ice–water. The combined aqueous solution is extracted with three 1-L portions of ethyl ether. The combined ether extracts are washed with 1 L of saturated brine and dried by stirring (magnetic stirring bar) over magnesium sulfate for 2 hr. The solution is filtered through a coarse, sintered-glass funnel and the ether is removed with a rotary evaporator at water aspirator pressure. After the pH of the residue is adjusted to 5 with a few drops of concentrated hydrochloric acid (Note 3), the residue is distilled to give 154–192 g (60–75%) of 2-hydroxybutanenitrile, bp 108–114°C (30 mm), as a colorless liquid (Note 4).
B. 2-[(1'-Ethoxy)-1-ethoxy]butanenitrile. A 1-L, three-necked, round-bottomed flask is equipped with a condenser topped with a calcium chloride drying tube, a magnetic stirring bar, a 500-mL pressure-equalizing addition funnel, and a thermometer. The flask is charged with 174 g (2.05 mol) of 2-hydroxybutanenitrile to which 0.5 mL of concentrated hydrochloric acid has been added. The addition funnel is charged with 221 g (3.07 mol) of ethyl vinyl ether (Note 5), which is then added dropwise to the stirring cyanohydrin at such a rate that the temperature is maintained at ca. 50°C. When the addition is complete, the mixture is heated to 90°C for 4 hr. The condenser is replaced with a distillation head and the dropping funnel and thermometer are replaced with stoppers. Direct distillation of the gold–yellow solution from the reaction flask yields 226–277 g (70–86%) of nearly pure 2-[(1'-ethoxy)-1-ethoxy]butanenitrile, bp 85–84°C (30 mm), as a colorless liquid (Note 6).
C. 2-Hydroxy-2-methylpentan-3-one. A dry, 5-L, three-necked (including a thermometer well), round-bottomed flask is equipped with a mechanical stirrer, low-temperature thermometer, nitrogen inlet, rubber septum, and a 1-L, graduated, pressure-equalizing addition funnel that is sealed with a rubber septum. The flask is charged with 775 mL of dry tetrahydrofuran (Note 7) and 166 g (1.64 mol) of dry diisopropylamine (Note 8). The contents of the flask are cooled to −10°C (dry ice–acetone bath) and 1095 mL (1.6 mol) of 1.5 M butyllithium in hexane (Note 9), which has been transferred to the addition funnel by means of a 16-gauge cannula and argon pressure, is slowly added to the stirring solution at such a rate as to maintain a temperature of −10°C. After the addition is complete, 50 mL of dry THF is added to the addition funnel with syringe to rinse the walls of the funnel; the rinse is added, and then the mixture is cooled to −75°C. The addition funnel is charged by syringe with 246 g (1.6 mol) of 2-[(1'-ethoxy)-1-ethoxy]butanenitrile, which is then added at such a rate that the temperature does not exceed −70°C. The mixture is stirred for 10 min and 104 g (1.8 mol) of dry acetone (Note 10) is added by syringe over a 30-min period at such a rate that the temperature of the reaction mixture does not exceed −70°C. When the addition is complete, the cooling bath is removed and the reaction mixture is allowed to warm to 0°C. The solution is poured into 1 L of water and the resulting mixture is concentrated at aspirator pressure with a rotary evaporator (30°C water bath) to remove the volatile organic compounds. The aqueous residue is extracted with three 1-L portions of methylene chloride. The organic extracts are combined and washed with two 500-mL portions of water and then concentrated with a rotary evaporator (25°C water bath) at aspirator pressure to obtain a yellow syrupy residue. This material is stirred with 680 mL of methanol and 340 mL of aqueous 5% sulfuric acid overnight at room temperature. The methanol is evaporated with a rotary evaporator (30°C water bath) at aspirator pressure and the yellow residue is extracted with three 1-L portions of ethyl ether. The combined ether extracts are shaken in a 4-L separatory funnel with 210 mL of 10 N aqueous sodium hydroxide for 15 min (Note 11). The layers are separated, and the ether layer is washed with 500 mL of brine and dried by stirring (magnetic stirring bar) over magnesium sulfate for 2 hr. The drying agent is removed by filtration through a coarse sintered-glass funnel and the ether is removed with a rotary evaporator (water bath below 40°C) at aspirator pressure. The yellow-orange liquid residue is distilled to obtain 82–115 g (45–63%) of 2-hydroxy-2-methylpentan-3-one, bp 57–65°C (15 mm), as a paleyellow liquid (Note 12) and (Note 13).
D. 2-Methyl-2-(trimethylsiloxy)pentan-3-one. A dry, 500 mL, three-necked, round-bottomed flask equipped with a mechanical stirrer, reflux condenser with a nitrogen inlet, and a thermometer is charged with 84 g (0.72 mol) of 2-hydroxy-2-methylpentan-3-one and 74 g (0.36 mol) of N,O-bis(trimethylsilyl)acetamide (Note 14). The mixture is heated at 100°C for 12 hr with stirring and then cooled to room temperature, at which point the mixture becomes a semisolid as the acetamide crystallizes. The semisolid mixture is diluted with 50 mL of water and stirred at room temperature for 1 hr (Note 15). After the stirring is stopped, 200 mL of hexane is added and the layers are separated. The aqueous layer is extracted with 100 mL of hexane. The combined hexane extracts are washed with four 100-mL portions of water and then dried over magnesium sulfate for 2 hr. After removal of the drying agent by filtration through a coarse sintered-glass funnel, the hexane is evaporated with a rotary evaporator (25°C water bath) at aspirator pressure. The crude, pale-yellow oil is distilled to afford 105–112 g (75–80%) of 2-methyl-2-(trimethylsiloxy)pentan-3-one, bp 71–75°C (15 mm), as a colorless liquid (Note 16).
2. Notes
1.
1. Caution! Sodium cyanide and 2-hydroxybutanenitrile are extremely toxic. Great care should be taken when using these materials. Reactions should be carried out in a well-ventilated hood and suitable protective clothing should be worn at all times.2.
Propanal was obtained from Aldrich Chemical Company and was used without further purification.
3.
If
HCl is omitted, the cyanohydrin reverts to
HCN and
propanal on attempted distillation. The checkers found it necessary to ensure that the residue was acidic by adjusting the pH to 5 by testing the residue with wet pH paper.
4.
The IR spectrum (neat) shows absorption at 3420, 2960, 2310, and 1460 cm
−1.
5.
Ethyl vinyl ether was obtained from Aldrich Chemical Company and was used without further purification.
6.
The IR spectrum (neat) shows absorption at 2970, 1425, and 1385 cm
−1. The C≡N absorption is not observed.
7.
Tetrahydrofuran is distilled under a
nitrogen atmosphere from
sodium–benzophenone immediately prior to use.
8.
Diisopropylamine is distilled under a
nitrogen atmosphere from
calcium hydride prior to use. It may be stored under
nitrogen for 1 week without redistillation.
9.
Concentrated butyllithium may ignite spontaneously on exposure to air or moisture. Manipulations with this reagent should be performed with care. The submitters used
butyllithium, 1.5 M in hexane from Foote Mineral Company, and measured it by transferring the solution to a
2-L, graduated cylinder stoppered with a rubber septum with a 15-gauge cannula and
argon. The solution was then transferred directly to the reaction vessel by the same procedure. The checkers used fresh
butyllithium, 1.55 M in hexane under argon, from Aldrich Chemical Company. Stainless steel cannulas with deflected points (double-tip syringe needles) are available from Ace Glass, Inc. and Aldrich Chemical Co.
10.
ACS Certified acetone was obtained from Fisher Chemical Company and distilled from Linde 3A molecular sieves immediately prior to use.
11.
Periodic shaking (once every 3 min) is sufficient to effect cyanohydrin hydrolysis.
12.
The
1H NMR (200 MHz, CDCl
3) spectrum is as follows δ: 1.12 (t, 3 H,
J = 7.2), 1.39 (s, 6 H), 2.59 (q, 2 H,
J = 7.2), 3.85 (s, 1 H). The IR spectrum (neat) shows absorption at 3450, 2960, and 1705 cm
−1.
13.
In one run, the checkers, at this point obtained
241 g, bp
116–120°C (12 mm) of the protected cyanohydrin (NMR), which had not been deprotected and hydrolyzed, rather than the expected product. In this case, the entire distillate was resubjected to the acid and base sequence, which afforded the desired product in a
61% overall isolated yield.
14.
N,O-Bis(trimethylsilyl)acetamide was obtained from Aldrich Chemical Company and used without further purification.
15.
This process is necessary to ensure hydrolysis of any unreacted
N,O-bis(trimethylsilyl)acetamide, which inevitably contaminates the product if the step is omitted.
16.
The
1H NMR spectrum (200 MHz, CDCl
3) is as follows: δ: 0.15 (s, 9 H), 1.02 (t, 3 H,
J = 7.2), 1.33 (s, 6 H), 2.67 (q, 2 H,
J = 7.2). The IR spectrum (neat) shows absorptions at 2980 and 1720 cm
−1.
3. Discussion
2-Methyl-2-(trimethylsiloxy)pentan-3-one (
1) is the prototype member of a series of α-trimethylsiloxy ketones that are useful for stereoselective aldol addition reactions (Equation 1).
2 β-Hydroxy ketones
2 may be converted into β-hydroxy acids,
2 β-hydroxy aldehydes,
3 and other β-hydroxy ketones.
4
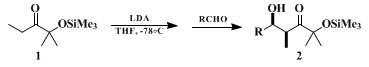
Compound
1 has also been prepared by the following methods. Addition of
ethylmagnesium bromide to the protected cyanohydrin of
acetone, followed by hydrolysis and silylation provides
1 in
40% yield (Equation 2).
2 Metallation of
1-methoxypropene by
butyllithium in
pentane5 gives
1-lithio-1-methoxypropene, which reacts with
acetone to give, after hydrolysis and silylation, ketone
1 in
25–30% overall yield (Equation 3).
6 The trimethylsilyl ether of acrolein
cyanohydrin, prepared by the method of Hünig,
7 may be metallated and added to
acetone to provide an enone which is hydrogenated to
1 (Equation 4).
8 Although the
overall yield in this sequence can be quite high, the intermediate enone
3 polymerizes very readily, and the procedure is not reliable on a large scale. Compound
1 has also been prepared by methylation of the lithium enolate of the lower homolog,
4, (Equation 5).
9 Although this alkylation provides
1 in
60% yield on a 2-mmol scale, the desired product is accompanied by a significant quantity of the dimethylated product, from which it is not easily separated.
8This preparation is referenced from:
Appendix
Chemical Abstracts Nomenclature (Collective Index Number);
(Registry Number)
brine
2-[(1'-Ethoxy)-1-ethoxy]butanenitrile
HCN
sulfuric acid (7664-93-9)
hydrochloric acid,
HCl (7647-01-0)
methanol (67-56-1)
ether,
ethyl ether (60-29-7)
sodium sulfite (7757-83-7)
sodium hydroxide (1310-73-2)
sodium cyanide (143-33-9)
propanal (123-38-6)
nitrogen (7727-37-9)
sodium bisulfite (7631-90-5)
acetone (67-64-1)
Benzophenone (119-61-9)
sodium (13966-32-0)
Pentane (109-66-0)
methylene chloride (75-09-2)
ethylmagnesium bromide (925-90-6)
magnesium sulfate (7487-88-9)
butyllithium (109-72-8)
Tetrahydrofuran,
THF (109-99-9)
hexane (110-54-3)
ethyl vinyl ether (109-92-2)
argon (7440-37-1)
calcium hydride (7789-78-8)
diisopropylamine (108-18-9)
2-methyl-2-(trimethylsiloxy)pentan-3-one,
3-Pentanone, 2-methyl-2-[(trimethylsilyl)oxy]- (72507-50-7)
2-Hydroxybutanenitrile (4476-02-2)
2-Hydroxy-2-methylpentan-3-one (2834-17-5)
1-methoxypropene (4188-68-5)
1-lithio-1-methoxypropene
N,O-bis(trimethylsilyl)acetamide
Copyright © 1921-, Organic Syntheses, Inc. All Rights Reserved