Org. Synth. 1990, 69, 129
DOI: 10.15227/orgsyn.069.0129
N-FLUOROPYRIDINIUM TRIFLATE: AN ELECTROPHILIC FLUORINATING AGENT
[Pyridinium, 1-fluoro-, salt with trifluoromethanesulfonic acid (1:1)]
Submitted by Teruo Umemoto
1, Kyoichi Tomita
1,2, and Kosuke Kawada
1,2.
Checked by Shlomo Rozen and Bruce E. Smart.
1. Procedure
Caution! Molecular fluorine is a very toxic and corrosive gas. The following reaction should be carried out in an efficient fume hood, and the experimenter should be familiar with the precautions necessary for safe handling of fluorine.3 Since molecular fluorine diluted with an inert gas is much safer to handle than pure fluorine, the use of a fluorinel nitrogen mixture comprising no more than 20% fluorine is recommended.
A.
N-Fluoropyridinium triflate (1). The reaction is carried out in the apparatus shown in
Figure 1. The
pressure regulator on the cylinder containing a mixture of 20% fluorine/80%
nitrogen (Note 1), and the
pressure gauge and
flowmeter on the
fluorine line are specifically designed for
fluorine service
(Note 2). The
fluorine and
nitrogen cylinders, pressure regulators, flowmeters,
valves, and
filters are connected with stainless-steel tubing. The Pyrex glass reaction vessel is connected to the metal tubing via Viton
® tubing, and the
fluorine gas is introduced into the vessel through a Pyrex glass tube (7 mm o. d.). The gas outlet from the reaction vessel is connected to a
granular alumina trap which consumes molecular
fluorine.
Figure 1. A—flowmeter (Matheson model 7825); B—flowmeter (Hastings model CST); C—alumina trap; D—reactor system; E—bubble counter containing perfluorotributylamine; F—pressure regulator (Matheson model 63–5512); G, I, K, O, P—stainless-steel values; H—pressure gauge (Matheson model 63–5512); J, N—stainless-steel filters; L—pressure regulator for nitrogen; M, Q, R, S—brass valves; T—Viton tubing; U—Teflon corks; V—Pyrex Claisen Adapter; W—Pyrex flask; X—Pyrex tube; Y—Teflon-coated stirring bar; Z— −40°C cooling bath.
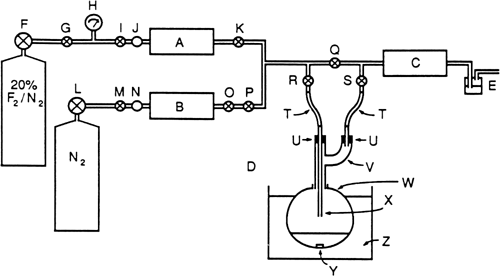
The 300-mL, round-bottomed reaction flask is charged with 4.74 g (0.06 mol) of pyridine (Note 3), 10.3 g (0.06 mol) of sodium triflate (Note 4), and 80 mL of dry acetonitrile (Note 5). The system is purged with nitrogen, and the reaction mixture is chilled and maintained at −40°C. The flow of dilute fluorine is started, and the flow rates from the nitrogen and fluorine cylinders are adjusted to introduce a 10% fluorine/90% nitrogen mixture at a rate of 90 mL/min just above the surface of the rapidly stirred solution (Note 6). When a total of 2.7 L of fluorine (0.12 mol) is introduced (Note 7), the flow of fluorine is discontinued and nitrogen only is flowed through the system at a rate of 45 mL/min for 30 min while keeping the reaction mixture at −40°C. The reaction mixture is then warmed to room temperature and filtered through a pad of Celite to remove the sodium fluoride. The filtrate is concentrated to dryness with a rotary evaporator without heating. The crystalline residue is washed with 30 mL of dry ethyl acetate to give 11.0–12.0 g (74–81%) of crude product, mp 178–181°C. The crude material is dissolved in 18 mL of dry acetonitrile at room temperature, and 36 mL of dry diethyl ether is added. The precipitated crystals are collected by filtration under nitrogen (Note 8) to give 10.0–10.3 g (68–70%) of pure N-fluoropyridinium triflate, mp 182°C (Note 9),(Note 10),(Note 11).
B. 3-Methoxy-17-trimethylsiloxy-1,3,5(10), 16-estratetraene. A 125-mL, two-necked round-bottomed flask equipped with a reflux condenser and a magnetic stirrer is purged with argon and charged with 6.8 g (0.024 mol) of estrone 3-methyl ether (Note 12), 50 mL of dry benzene, and 4.0 mL (2.9 g, 0.029 mol) of triethylamine. The solution is stirred, 4.9 mL (5.6 g, 0.025 mol) of trimethylsilyl triflate (Note 13) is added through a syringe, and the mixture is refluxed for 1.5 hr. The reaction mixture is allowed to cool to room temperature, whereupon it separates into two layers. Dry hexane (40 mL) is added, and the upper hexane–benzene layer is separated, washed successively with saturated sodium bicarbonate and water, and then dried over magnesium sulfate. The drying agent is removed by filtration, and the filtrate is transferred to a 125-mL, round-bottomed, tared flask. The solution is evaporated to a constant weight with a rotary evaporator, initially at water-aspirator pressure and then at 0.5–1 mm, to leave 8.6 g (100%) of pale-yellow enol trimethylsilyl ether. This material is used immediately in Part C without purification (Note 14).
C. 16α-Fluoro-3-methoxy-1,3,5(10)-estratrien-17-one (16α-fluoroestrone 3-methyl ether). The 125-mL, round-bottomed flask containing the enol silyl ether from Part B is purged with argon, and 50 mL of dry dichloromethane is added. N-Fluoropyridinium triflate (1) (6.5 g, 0.026 mol) is added in one portion, and the mixture is stirred at 20–25°C for 8 hr (Note 15). The reaction mixture is poured into water and extracted with three 60-mL portions of dichloromethane. The combined organic extracts are washed with saturated sodium bicarbonate and then with water, and dried over magnesium sulfate. The drying agent is removed by filtration and the solution is evaporated to dryness with a rotary evaporator. The resulting pale-yellow solid is column-chromatographed on silica gel (250 g, 60 × 4.5-cm column) using dichloromethane eluant (Note 16) to give 950 mg (14%) of estrone 3-methyl ether starting material and 4.8 g (66%) of 16α-fluoroestrone 3-methyl ether as a colorless solid, mp 157°C (Note 17),(Note 18),(Note 19).
2. Notes
1.
A cylinder containing 20%
fluorine/80%
nitrogen was obtained from Air Products & Chemicals, Inc.
2.
The checkers used a
Matheson model B15F-679 single-stage pressure regulator, a
model 63-5512 pressure gauge, and a
model 7825 flowmeter. Information on equipment designed to handle
fluorine can be found in the bulletin Tech-Brief TB-115, published by Matheson Gas Products.
3.
Anhydrous
pyridine (≥99%) packaged under nitrogen was purchased from Aldrich Chemical Company, Inc., and used without further purification.
4.
Sodium trifluoromethanesulfonate (
triflate) was prepared from
trifluoromethanesulfonic acid (Aldrich Chemical Company, Inc.) as follows. A solution of
26.5 g (0.66 mol) of sodium hydroxide in 50 mL of water was added dropwise to
100 g (0.67 mol) of triflic acid chilled in an
ice bath. The solution was concentrated to dryness with a rotary evaporator, and the residual solid was recrystallized from
65 mL of acetonitrile. The collected solid is dried at 80°C under vacuum for 24 hr to give
90 g of pure
sodium triflate.
5.
Acetonitrile was distilled from
calcium hydride under
nitrogen immediately before use.
6.
A powerful magnetic stirrer was used to obtain a stirring rate of about 80 rps. The checkers also used a VIBRO-Mixer E1 (Chemapec, Inc.). The checkers found that the yield was unaffected if the
fluorine is introduced below rather than above the surface of the solution.
7.
A substantial excess of
fluorine over the theoretical, equimolar amount is needed to complete the reaction because of the low solubility of
fluorine. The amount of
fluorine required can vary depending on the scale of reaction, the flow rate, and the efficiency of mixing.
8.
The submitters carried out the filtration procedure in air. Filtration in wet air should be avoided.
9.
The submitters report obtaining
13.2 g of crude product, mp
181–184°C, and
12.0 g (
81%) of recrystallized material, mp
185–187°C.
10.
The product obtained by the checkers is pure by NMR analyses.
N-Fluoropyridinium triflate (1) has the following spectral properties:
1H NMR (CD
3CN) δ: 8.32 (m, 2 H), 8.77 (m, 1 H), 9.33 (dd, 2 H,
J = 16.7);
19F NMR (CD
3CN) δ: 48.8 (bs, 1 F, N-F), −77.6 (s, 3 F, CF
3): IR (Nujol on NaCl plate) cm
−1: 3140 (s), 3120 (s), 3080 (s), 3050 (s), 1600 (m), 1485 (s), 1475 (s), 1330 (w), 1270 (s), 1250 (s), 1220 (s), 1200 (s), 1175 (s), 1160 (s), 1090 (m), 1055 (w), 1020 (s), 805 (m), 770 (s), 755 (m), 630 (s).
11.
N-Fluoropyridinium triflate is stable and can be stored indefinitely under a dry atmosphere. It slowly decomposes in water. The submitters report that it has a half-life of 13 days in D
2O at room temperature.
12.
Estrone 3-methyl ether [3-methoxyestra-1,3,5(10)-trien-17-one] was purchased from Sigma Chemical Company.
13.
Trimethylsilyl triflate was obtained from Aldrich Chemical Company, Inc. and used without further purification.
14.
The product exhibits the following partial spectral data:
1H NMR (CDCl
3) δ: 0.21 (s, 9 H, CH
3Si), 0.87 (s, 3 H, 18-CH
3), 3.77 (s, 3 H, OCH
3), 4.53 (m, 1 H, 16-H). This silyl enol ether is sensitive to hydrolysis, and the submitters recommend that it be isolated in the same flask that is used for its subsequent reaction in Part C.
15.
Crystals of
1 gradually disappear as the reaction proceeds, and the mixture turns orange and finally becomes homogeneous when the reaction is completed.
16.
Each fraction was monitored by thin-layer chromatography on
silica gel (Merck Silica Gel 60 F-254). The
Rf values (
dichloromethane) of the product and starting
estrone 3-methyl ether are 0.53 and 0.40, respectively.
17.
The product has the following characteristic spectral properties:
1H NMR (360 MHz, CDCl
3) δ: 0.95 (s, 3 H, 18-CH
3), 3.77 (s, 3 H, OCH
3), 5.13 (dd, 1 H,
J = 50.6, 7.3, 16 β-H), 6.64 (d, 1 H,
J = 2.7, 4-H), 6.72 (dd, 1 H,
J = 8.6, 2.7, 2-H), 7.19 (d, 1 H,
J = 8.6, 1-H);
19F NMR (CDCl
3) δ: −192.7 (m); IR (KBr) cm
−1: 2900, 2850, 1750, 1600, 1500, 1460, 1440, 1310, 1240, 1030, 1000: MS
m/e (relative intensity) 304 (2.7), 303 (21.5), 302 (M
+) (100), 301 (3.7).
18.
The product contains about
4% of the
16β-fluoroestrone epimer;
1H NMR (360 MHz, CDCl
3) δ: 4.76 (dt,
J = 50, 8; 16α-H);
19F NMR (CDCl
3) δ: −185.3 (dd,
J = 50, 22; 16β-F).
19.
Identical yields of recovered starting material and product were obtained when Parts B and C were run on 2.5 times the scale. The submitters report obtaining a
78% yield of product, mp
145–149°C (recrystallized from
ethyl acetate/
hexane after chromatography) containing a small but unspecified amount of its epimer, along with
11% recovered starting material and
12% 2-pyridyl triflate, which is a decomposition product of
1. Anal. calcd. for C
19H
23O
2F: C, 75.47; H, 7.66. Found: C, 75.52; H, 7.81.
3. Discussion
Electrophilic fluorinating agents such as F
2,
4 CF
3OF,
5 FClO
3,
6 CF
3COOF,
7 CH
3COOF,
8 XeF
2,
9 and CsSO
4F
10 require the use of special equipment and techniques because of their explosive, toxic, unstable, or hygroscopic nature.
N-Fluoroperfluoropiperidine,
11 N-fluoropyridone,
12 and
N-fluoro-
N-alkyltoluenesulfonamides
13 are easy to handle, but their low reactivity limits the scope of their applications. Recently, a variety of fluorinating agents have been developed;
N-fluoroquinuclidinium salts,
14 N-fluorobis[(trifluoromethyl)sulfonyl]imide,
15 N-fluorosultams,
16 N-fluoro-3,3-dimethyl-2,3-dihydro-1,2-benzothiazole 1,1-dioxide,
17 perfluoro-[N-fluoro-N-(4-pyridyl)methanesulfonamide],
18 N-fluoro-o-benzenedisulfonimide,
19 N-fluorobenzenesulfonimide,
20 and
N-fluorotriethylenediammonium salts.
21N-Fluoropyridinium trifluoromethanesulfonate (
triflate) and its analogs are widely applicable, stable fluorinating agents with varying degrees of power and selectivity in fluorinations.
22 23 24 25 26 27 28 29 30 31 The procedure given here represents a general method for preparing substituted
N-fluoropyridinium triflates.
N-Fluoropyridinium salts with a different counter anion such as BF
4, PF
6, or ClO
4 can be prepared in good yields by the same method. However, depending on the ring substituents or counteranions, the modified or other methods should be adopted.
24,25 Counteranion-bound
N-fluoropyridinium salts
6 and
7 are prepared in good yields by fluorination of the corresponding pyridinesulfonic acids with 10% F
2/N
2 in aqueous
acetonitrile at −20°C.
24,25
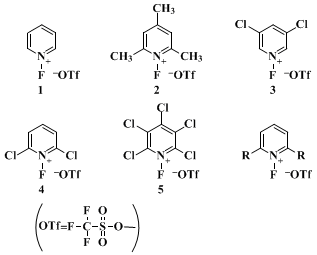
The reactivity of the
N-fluoropyridinium salts can be adjusted by varying the substituents on the
pyridine ring. Triflates
1–5, whose fluorinating power increases in the order
2 <
1 <
3 <
4 <
5, are the most generally useful reagents. Salts
6 and 7, power order
6 <
7, are the highly selective reagents. Other useful
N-fluoropyridinium salts were described in the paper.
23 The reagents are all stable, crystalline materials and thus can be handled routinely. However, it should be noticed that the relative stability of the
N-fluropyridinium salts decreases with increasing fluorinating power. Examples of fluorinations which illustrate their use are given in Table I. The weakest reagent,
2, is most suited for fluorinating reactive or easily oxidized compounds, such as carbanions, enamines, and sulfides, whereas the most potent reagents
4 and
5 are preferred for fluorinating alkenes and aromatic rings.
Salt 1 of intermediate reactivity is effective with moderately electron-rich substrates, such as enol alkyl ethers, enol
silyl ethers, and activated
vinyl acetates.
Salt 3 is useful with activated aromatic compounds but an elevated reaction temperature is required.
TABLE I
ELECTROPHILIC FLUORINATIONS WITH N-FLUOROPYRIDINIUM TRIFLATES
|
|
Substrate
|
Reagenta
|
Conditions
|
Product
|
Yield (%)b
|
|
|
n-C12H25MgCl
|
2
|
0°C, 30 min in Et2O
|
n-C12H25F
|
75c
|
|
NaCH(COOEt)2
|
2
|
0°C, 0.1 hr in THF
|
CHF(COOEt)2
|
73
|
|
CH2(COOEt)2
|
2d
|
AlCl3,e 80°C 24 hr in CH2ClCH2Cl
|
CF2(COOEt)2
|
76f
|
CHF(COOEt)2
|
19f
|
|
p-ClC6H4SCH3
|
2
|
RT, 8 hr in CH2Cl2
|
p-ClC6H4SCH2F
|
76
|
|
|
2
|
(1) −15°C, 1 hr in CH2Cl2/CH3CN(1/4)
|
|
54
|
1
|
(2) RT, ca. 12 hr in conc. HCl/DMF
|
46
|
|
|
1
|
RT, 7 hr in CH2Cl2
|
|
87c
|
|
|
1
|
60°C, 30 min in CH2ClCH2Cl
|
|
63f
|
|
|
1
|
Reflux, 7 hr in CH2Cl2
|
|
86h
|
|
|
1
|
Reflux, 10 hr in CH2Cl2
|
|
71f
|
|
PhH
|
5
|
Reflux, 2 hr in CH2Cl2
|
Ph-F
|
48f
|
|
PhOH
|
4
|
RT, 5 hr in CH2Cl2
|
F-C6H4OH (o:p)
|
49c,j (1.3:1)
|
|
PhOH
|
6
|
Reflux, 1.5 hr in CHCl2CH2Cl
|
o-F-C6H4OH
|
45c,j
|
|
PhNHCOOEt
|
3
|
Reflux, 5.5 hr in CH2ClCH2Cl
|
F-C6H4NHCOOEt (o:p)
|
51j (2.6:1)
|
|
PhNHCOOEt
|
7
|
Reflux, 72 hr in CH2ClCH2Cl
|
F-C6H4NHCOOEt (o:p)
|
66j (12:1)
|
|
PhCH=CH2
|
5
|
RT, 1 hr in AcOH
|
PhCH(OAc)CH2F
|
72
|
|
aEquimolar N-fluoropyridinium triflate unless noted otherwise.
|
bIsolated yields unless noted otherwise. Yield calculations based on the used amounts of triflates.
|
|
|
|
|
e0.4 Equivalents of AlCl3.
|
|
|
g1.5 Equivalents of dihydropyran.
|
|
|
|
|
jA considerable amount of the starting substrate (16–38%) was recovered.
|
|
|
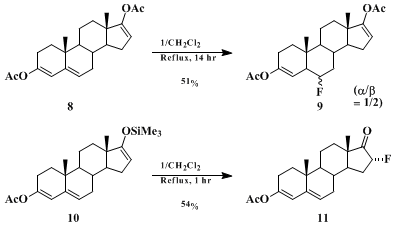
N-Fluoropyridinium triflate shows high regioselectivity in its fluorinations, as evidenced by the results in Schemes 1 and 2. With steroids 8 and 10, each having two reactive sites, 1 reacts to give exclusively the 6-fluoro steroid 9 and the 16-fluoro steroid 11, respectively. Thus 1 can distinguish between a conjugated and a nonconjugated vinyl acetate, and between an enol silyl ether and a conjugated vinyl acetate in its fluorinations. The present procedure for converting estrone enol silyl ether to 16α-fluoroestrone also shows that 1 selectively reacts with an enol silyl ether moiety in the presence of an activated aromatic ring.
The fluorination of
12, easily prepared from the corresponding triketo steroid, with an equimolar amount of
1 (Scheme 2) shows the remarkable ability of
1 to distinguish disubstituted from trisubstituted enol
silyl ethers. The
9α-fluoro steroid
13 is produced in 51% yield (78% based on recovered triketo steroid) and the combined yield of the other products is only 4.6%.
23 It thus is apparent that
1 reacts almost exclusively with the trisubstituted enol ether moiety. The new, selective direct fluorination at the 9α-position holds considerable promise as a means to prepare biologically important
9α-fluoro steroids.
32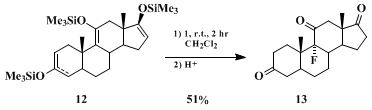
Salt
6 or
7 affords exclusive or greatly preferential ortho-fluorination of
phenol and
aniline derivatives (Table I). Some fluorinations may be stereoselective. When allowed to react with 3,
17β-diacetoxy-3,5-androstadiene,
1 gives a 1 : 2 mixture of α- and β-fluorination at 6-position, while the bulky salt
2 affords a 1 : 8.5 mixture of them, resulting from less hindered fluorination.
23 A diastereoselective fluorination of a chiral enolate with salt
2 was reported.
30With the increasing importance of organofluorine compounds in the development of new materials, pharmaceuticals, and agricultural chemicals, N-fluoropyridinium salts should find extensive use as mild and selective fluorinating agents.
Appendix
Chemical Abstracts Nomenclature (Collective Index Number);
(Registry Number)
fluorinel nitrogen
N-fluoro-o-benzenedisulfonimide
N-fluorobenzenesulfonimide
N-fluorotriethylenediammonium
9α-fluoro steroids
Pyridinium, 1-fluoro-, salt with trifluoromethanesulfonic acid (1:1)
Benzene (71-43-2)
ethyl acetate (141-78-6)
diethyl ether (60-29-7)
aniline (62-53-3)
acetonitrile (75-05-8)
sodium hydroxide (1310-73-2)
sodium bicarbonate (144-55-8)
phenol (108-95-2)
Salt (7647-14-5)
nitrogen (7727-37-9)
pyridine (110-86-1)
dichloromethane (75-09-2)
magnesium sulfate (7487-88-9)
sodium fluoride (7681-49-4)
vinyl acetate (108-05-4)
dihydropyran
hexane (110-54-3)
vinyl (2669-89-8)
triethylamine (121-44-8)
argon (7440-37-1)
calcium hydride (7789-78-8)
9α-fluoro
Fluorine (7782-41-4)
triflate
trifluoromethanesulfonic acid,
triflic acid (1493-13-6)
silyl (13765-44-1)
trimethylsilyl ether (107-46-0)
silyl ether (13597-73-4)
trimethylsilyl triflate (27607-77-8)
N-fluoropyridinium,
N-fluropyridinium
sodium triflate,
Sodium trifluoromethanesulfonate (2926-30-9)
perfluorotributylamine (311-89-7)
estrone 3-methyl ether,
3-methoxyestra-1,3,5(10)-trien-17-one
16α-Fluoro-3-methoxy-1,3,5(10)-estratrien-17-one,
16α-fluoroestrone 3-methyl ether (2383-28-0)
16β-fluoroestrone
2-pyridyl triflate (65007-00-3)
estrone enol silyl ether
16α-fluoroestrone
17β-diacetoxy-3,5-androstadiene
N-Fluoropyridinium triflate,
N-Fluoropyridinium trifluoromethanesulfonate (107263-95-6)
N-Fluoroperfluoropiperidine (836-77-1)
N-fluoropyridone
N-fluoroquinuclidinium
N-fluoro-3,3-dimethyl-2,3-dihydro-1,2-benzothiazole 1,1-dioxide (124170-23-6)
perfluoro-[N-fluoro-N-(4-pyridyl)methanesulfonamide]
3-Methoxy-17-trimethylsiloxy-1,3,5(10), 16-estratetraene
Copyright © 1921-, Organic Syntheses, Inc. All Rights Reserved