Org. Synth. 1990, 69, 212
DOI: 10.15227/orgsyn.069.0212
A GENERAL SYNTHETIC METHOD FOR THE OXIDATION OF PRIMARY ALCOHOLS TO ALDEHYDES: (S)-(+)-2-METHYLBUTANAL
[Butanal, 2-methyl-, (S)-]
Submitted by Pier Lucio Anelli, Fernando Montanari, and Silvio Quici
1.
Checked by Katsumasa Nonoshita and Hisashi Yamamoto.
1. Procedure
A 1-L, three-necked, round-bottomed flask is fitted with a mechanical stirrer, pressure-equalizing dropping funnel, and a thermometer. The flask is charged with 44.05 g (0.50 mol) of (S)-(−)-2-methyl-1-butanol (Note 1), 0.78 g (5 mmol) of 2,2,6,6-tetramethylpiperidin-1-oxyl (Note 2), 170 mL of dichloromethane, and a solution of 5.95 g (0.050 mol) of potassium bromide in 25 mL of water (Note 3). The reaction mixture is vigorously stirred and cooled to −10°C with a salt–ice bath, then 550 mL (0.55 mol) of 1 M aqueous sodium hypochlorite (Note 4) at pH 9.5 (Note 5) is added over 15–20 min (Note 6), keeping the temperature of the reaction mixture between 10 and 15°C. The mixture is stirred for a further 3 min (Note 7). The orange organic phase is separated and the aqueous phase (Note 8) is extracted with 50 mL of dichloromethane. The combined organic extracts are washed with 100 mL of 10% aqueous hydrochloric acid containing 1.6 g (0.010 mol) of potassium iodide (Note 9), 60 mL of 10% aqueous sodium thiosulfate (Note 10), and 60 mL of water (Note 11). The organic phase is dried over anhydrous magnesium sulfate and then distilled at atmospheric pressure through a 20-cm Vigreux distilling column to give 35.3–36.3 g (82–84%) (Note 12) of (S)-(+)-2-methylbutanal as a colorless oil, bp 90–92°C (GC purity >99%) (Note 13), [α]D22 +36.8° (acetone, c 2.5) (Note 14),(Note 15),(Note 16).
2. Notes
1.
(S)-(−)-2-Methyl-1-butanol (GC purity >99.5%);
[α]D20 −6.6±0.3° (
ethanol,
c 10) was purchased from Fluka Chemie AG. Esterification with
(R)-(+)-3,3,3-trifluoro-2-methoxy-2-phenylpropionic acid (Mosher's acid),
2 and subsequent
1H and
19F NMR analyses at 300 MHz of the resulting ester showed an enantiomeric purity of
(S)-(−)-2-methyl-1-butanol >99%.
2.
2,2,6,6-Tetramethylpiperidin-1-oxyl from Nacalai Tesque, Inc., Kyoto, Japan, also available from Janssen Chimica, Beerse, Belgium, was used.
4-Methoxy-2,2,6,6-tetramethylpiperidin-1-oxyl, prepared according to the procedure of Endo,
3 can also be used.
43.
In the absence of
potassium bromide longer reaction times are required.
44.
Concentrations of aqueous
sodium hypochlorite in the range 0.3–2.0
M have been used successfully.
5.
The pH (~12.7) of fresh commercial
1 M aqueous sodium hypochlorite is adjusted to 9.5 by dissolving
17 g of sodium hydrogen carbonate per liter immediately before use.
6.
If the reaction is carried out on a 1–10-mmol scale, the temperature is easily maintained at 0°C and the reaction is over in a few minutes.
4 On a larger scale, a very efficient cooling system is required to maintain the temperature at about 0°C. When conventional laboratory equipment is used, the conditions described in this procedure are a reasonable compromise between two requirements: (a) fast addition of the aqueous
sodium hypochlorite and (b) temperature in the reaction medium low enough to minimize the catalyst decomposition.
4 Longer reaction times increase slightly the formation of
2-methylbutanoic acid.
7.
At this stage the reaction can be monitored by GC: 1 m × 3 mm OV 101 5% on Chromosorb HP 100–120-mesh column, 50°C (2 min), then 50–90°C (15°C/min).
8.
(S)-(+)-2-Methylbutanoic acid,
[α]D20 +18.7° (
ethanol,
c 1.1) (lit.
5 [α]D22 +16.3°,
ethanol,
c 1.1) (
1.5–2.6 g,
3–5% yield) can be isolated by acidic workup of the aqueous phase.
9.
Washing with
hydrochloric acid and
potassium iodide removes
2,2,6,6-tetramethylpiperidin-1-oxyl from the organic phase.
6 Because of its volatility, the catalyst cannot be eliminated in the distillation of crude aldehyde.
10.
Washing with
10% aqueous sodium thiosulfate leads to a colorless organic phase, indicating total elimination of the catalyst.
11.
The aqueous phase must be neutral. Acidic impurities catalyze trimerization of the anhydrous aldehyde
7 8 in the distillation stage.
12.
Yields can be further increased with a more efficient separation of the
(S)-(+)-2-methyl-1-butanal contained in the top fractions.
13.
The spectral properties of
(S)-(+)-2-methylbutanal are as follows: IR (film) cm
−1: 2970, 2940, 2890, 2820, 2710, 1725, 1460;
1H NMR (300 MHz, CDCl
3) δ: 0.94 (t, 3 H), 1.09 (d, 3 H), 1.33–1.54 (m, 1 H), 1.64–1.85 (m, 1 H), 2.18–2.33 (m, 1 H), 9.63 (d, 1 H).
14.
Reduction with
borane/tetrahydrofuran9 regenerated enantiomerically pure
(S)-(−)-2-methyl-1-butanol, as shown by esterification with
Mosher's acid and subsequent NMR analysis of the ester (see
(Note 1).
15.
When practical,
(S)-(−)-2-methyl-1-butanol (GC purity 95%);
[α]D20 −6.3 ± 0.5° (EtOH,
c 10) from Fluka Chemie was used, and
(S)-(+)-2-methylbutanal having
[α]D20 +33.1° (
acetone,
c 2.5) was obtained.
16.
Oxidation of
(S)-(−)-2-methyl-1-butanol to
(S)-(+)-2-methylbutanal has been previously carried out in low yields by
chromium oxidation,
10 under phase-transfer catalysis,
11 or by Swern oxidation in the presence of
tributylamine.
12
3. Discussion
Oxammonium salts
1 have been used extensively either in stoichiometric or in catalytic amounts
13 for the oxidation of primary and secondary alcohols to the corresponding carbonyl derivatives.
The catalytic procedure described here allows a fast, cheap, and highly selective conversion of primary alcohols into aldehydes, using
sodium hypochlorite as the oxidant in a two-phase (
dichloromethane–water) system. Aqueous
sodium hypochlorite is buffered at pH 8.6–9.5 to ensure the presence of
hypochlorous acid in the organic layer.
14Oxammonium salt
1, the effective oxidant species, is continuously generated from nitroxyl radical
2 by
hypochlorous acid in the organic phase. Radical
2 is one of the most stable radicals known, and is easily prepared from the inexpensive triacetoneamine
3.15 16 17 18 The oxidation is very exothermic; for this reason scale-up of the reaction needs a very efficient cooling system to maintain the temperature in the optimum 0–15°C range. One one-hundredth (0.01) molar equivalent of nitroxyl radical
2 is generally used, but on this reaction scale the amount of catalyst can be reduced to 0.002 molar equivalent, without substantially affecting the reaction time.
Sodium hypochlorite is used in only slight excess and is entirely consumed, an unusual occurrence for reactions carried out under aqueous, organic two-phase conditions.
19 20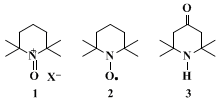
Conversion of saturated, primary alkyl and aryl alkyl alcohols into the corresponding aldehydes can be achieved by this method provided that the alcohols are entirely dissolved in the organic phase. Relatively unstable protective groups are not affected, as in the oxidation of the
acetonide of 1,2,6-hexanetriol, whereas conjugated and isolated double bonds give rise to side reactions that considerably decrease selectivities and yields.
4 Some examples of aldehydes synthesized with this method are reported in Table I. Under the same conditions, secondary alcohols are oxidized to ketones. Addition of catalytic amounts of quaternary onium salts allows fast and total conversion of primary alcohols and aldehydes into carboxylic acids making this methodology very versatile.
4
TABLE I
OXIDATION OF PRIMARY ALCOHOLS TO ALDEHYDES
|
|
Alcohol
|
Isolated Yield (%)
|
|
|
1-Heptanol
|
88
|
|
1-Octanol
|
92
|
|
1-Nonanol
|
924
|
|
1-Undecanol
|
934
|
|
|
85
|
|
Benzyl alcohol
|
904
|
|
p-Nitrobenzyl alcohol
|
89
|
|
m-Nitrobenyl alcohol
|
88
|
|
|
75
|
|
When the limitations outlined above are considered, the procedure described here appears to be easier and cheaper than most methods in the condensed phase known to date.
21 Furthermore, alkali halides are almost the only contaminants in the waste water, making the scale up of this method very attractive.
The conditions reported for the preparation of
S-(+)-2-methylbutanal also have been used for the oxidation of diols containing both primary and secondary hydroxyl groups into carbonyl derivatives; for example,
1,10-undecanediol can be converted into
10-hydroxyundecanal, or
10-oxoundecanal, or
10-oxoundecanoic acid depending on the amount of
NaOCl used and on the presence of a phase-transfer catalyst.
22Very hydrophilic alcohols are conveniently oxidized under solid–liquid two-phase conditions (LiOCl–CH
2Cl
2) in the presence of
NaHCO3 and 0.01 molar equiv of
2,2,6,6-tetramethylpiperidin-1-oxyl or its 4-methoxy derivative.
22 Commercial solid LiOCl contains 7% of water so that the reaction conditions can be assimilated to those of a pseudo solid–liquid system.
23Under these conditions 1-butanol is converted into butanal in 80% yield in 20 min at 0°C, whereas the hydrophilic 1,4-butanediol and 1,5-pentanediol are readily oxidized into the corresponding lactones.
24
Appendix
Chemical Abstracts Nomenclature (Collective Index Number);
(Registry Number)
(S)-(−)-2-methyl-1-butanol
(R)-(+)-3,3,3-trifluoro-2-methoxy-2-phenylpropionic acid (Mosher's acid)
Mosher's acid
acetonide of 1,2,6-hexanetriol
m-Nitrobenyl alcohol
alcohol,
ethanol (64-17-5)
hydrochloric acid (7647-01-0)
sodium hydrogen carbonate,
NaHCO3 (144-55-8)
potassium iodide (7681-11-0)
sodium thiosulfate (7772-98-7)
1-butanol (71-36-3)
acetone (67-64-1)
Benzyl alcohol (100-51-6)
potassium bromide (7758-02-3)
butanal (123-72-8)
hypochlorous acid (7790-92-3)
1,5-Pentanediol (111-29-5)
1-heptanol (111-70-6)
sodium hypochlorite,
NaOCl (7681-52-9)
dichloromethane (75-09-2)
2-methylbutanoic acid (600-07-7)
magnesium sulfate (7487-88-9)
chromium (7440-47-3)
borane (7440-42-8)
1-Octanol (111-87-5)
Tetrahydrofuran (109-99-9)
1,4-butanediol
tributylamine (102-82-9)
p-Nitrobenzyl alcohol (619-73-8)
2,2,6,6-tetramethylpiperidin-1-oxyl (2564-83-2)
4-Methoxy-2,2,6,6-tetramethylpiperidin-1-oxyl (95407-69-5)
1,10-undecanediol
10-hydroxyundecanal
10-oxoundecanal
10-oxoundecanoic acid
1-Nonanol (143-08-8)
1-Undecanol (112-42-5)
(S)-(+)-2-METHYLBUTANAL,
Butanal, 2-methyl-, (S)-,
(S)-(+)-2-methyl-1-butanal,
S-(+)-2-methylbutanal (1730-97-8)
(S)-(+)-2-Methylbutanoic acid
Copyright © 1921-, Organic Syntheses, Inc. All Rights Reserved