Org. Synth. 1987, 65, 119
DOI: 10.15227/orgsyn.065.0119
GEMINAL ACYLATION-ALKYLATION AT A CARBONYL CENTER USING DIETHYL N-BENZYLIDENEAMINOMETHYLPHOSPHONATE: 2-METHYL-2-PHENYL-4-PENTENAL
[4-Pentenal, 2-methyl-2-phenyl-]
Submitted by Steven K. Davidsen, Gerald W. Phllips, and Stephen F. Martin
1.
Checked by Robert G. Aslanian, Max Tishler, and Andrew S. Kende.
1. Procedure
A.
N-Bromomethylphthalimide (2). A
1-L, three-necked, round-bottomed flask is fitted with a
mechanical stirrer, a
125-mL pressure-equalizing dropping funnel, and a
thermometer. The flask is charged with
50.0 g (0.28 mol) of N-hydroxymethylphthalimide (Note 1) and
200 mL of 48% aqueous hydrobromic acid (Note 2). The flask is immersed in an
ice bath, and
75 mL of concentrated sulfuric acid is added with stirring over a period of about 15 min
(Note 3). On completion of the addition, the flask is removed from the ice bath, heated at 60–70°C for 5 hr, and then cooled overnight in a
refrigerator. The solid is collected by suction filtration using a
125-mm glass funnel with a coarse frit. The crude product is washed thoroughly with three 100-mL portions of cold water, two
50-mL portions of cold 10% aqueous ammonium hydroxide, and finally with three 100-mL portions of cold water
(Note 4). The crude product thus obtained is completely dried under reduced pressure at room temperature over
phosphorus pentoxide to give
57.1–63.8 g (
85–95%) of
N-bromomethylphthalimide as a light-tan solid, mp
142–147°C. Although the material thus obtained may be used in the next step without further purification, it may also be recrystallized from dry
acetone, mp
147–148°C (lit. mp
148°C,
2 148–149°C3)
(Note 5).
B.
Diethyl phthalimidomethylphosphonate (3). A
500-mL, one-necked, round-bottomed flask equipped with a
magnetic stirring bar and an
efficient reflux condenser (ca. 80 cm long) is charged with
51.2 g (0.21 mol) of dry N-bromophthalimide (2) and
43.1 g (0.26 mol) of freshly distilled triethyl phosphite (Note 6). The mixture is immersed in an
oil bath and the temperature of the oil bath gradually increased over about 15 min to 85–100°C, whereupon the solid dissolves and a vigorous, exothermic reaction ensues
(Note 7). When the reaction has subsided, the oil bath is lowered and the condenser is removed. The flask is fitted for simple distillation, and
ethyl bromide and other volatile materials are distilled from the reaction mixture over a period of 3–4 hr by continued heating at 115°C (oil bath). The resulting light-yellow oil is cooled to room temperature, whereupon it solidifies
(Note 8). The crude product is collected by suction filtration and washed with three
100-mL portions of cold petroleum ether (bp
60–68°C) to give
50.1–56.3 g (
80–90%) of
diethyl phthalimidomethylphosphonate as white crystals, mp
60–63°C, which are used in the next step without further purification. Recrystallization of this material from
diethyl ether/
petroleum ether (bp
60–68°C) affords pure
3, mp
65–67°C (lit.,
3 67°C)
(Note 9).
C.
Diethyl N-benzylideneaminomethylphosphonate (5). A
2-L, two-necked, round-bottomed flask is equipped with a mechanical stirrer and a 125-mL pressure-equalizing dropping funnel fitted with a
calcium chloride drying tube. The flask is charged with
50.0 g (0.17 mol) of diethyl phthalimidomethylphosphonate (3) dissolved in
750 mL of absolute ethanol (Note 10). To this solution is then added
6.4 g (0.19 mol) of 95% hydrazine (Note 11) in
50 mL of absolute ethanol, and the resulting mixture is stirred overnight at room temperature. The dropping funnel is replaced with a reflux condenser bearing a calcium chloride drying tube, and the mixture is heated at reflux for 4 hr and then cooled to 0–5°C in an ice bath. The precipitated
phthalhydrazide is collected by suction filtration and thoroughly washed with three
125-mL portions of benzene. The excess solvents and
hydrazine are completely removed under reduced pressure on a
rotary evaporator and then under high vacuum (0.1 mm)
(Note 12) and
(Note 13). The crude
diethyl aminomethylphosphonate (4)4,5 (Note 14) thus obtained is dissolved in
350 mL of reagent-grade benzene, and the solution is cooled overnight in the refrigerator. Any additional
phthalhydrazide that precipitates is removed by suction filtration and washed with two
25-mL portions of benzene. The filtrate and washings are combined in a
1-L, one-necked flask equipped with magnetic stirring bar, and the solution is cooled to 5–10°C, at which time
21.2 g (0.20 mol) of freshly distilled benzaldehyde is added in one portion with stirring. The mixture is stirred for 4 hr at room temperature, the flask is fitted with a
Dean–Stark trap and a reflux condenser and heated overnight at reflux with constant removal of water. The solution is cooled to approximately room temperature, and the excess solvents are removed under reduced pressure. The crude product is purified by vacuum distillation, bp
145–149°C (0.05 mm), to give
37.5–40.2 g (
84–90%) of pure
diethyl N-benzylideneaminomethylphosphonate (5)6,7 as a light-yellow oil
(Note 15).
D. 2-Methyl-2-phenyl-4-pentenal (7). A dry, 100-mL, three-necked, round-bottomed flask with 14/20 joints is fitted with a magnetic stirrer, a reflux condenser, and a rubber septum (Note 16). The flask is charged with 50 mL of anhydrous tetrahydrofuran (Note 17) and cooled to −78°C in a dry ice–isopropyl alcohol bath, and a solution of butyllithium (12.0 mmol) in hexane (Note 18) is added with stirring. To this stirred solution is added dropwise via syringe a solution of 3.06 g (12.0 mmol) of diethyl N-benzylideneaminomethylphosphonate (5) in 5 mL of anhydrous tetrahydrofuran, and the colored solution is stirred an additional hour at −78°C. A solution containing 1.20 g (10.0 mmol) of freshly distilled acetophenone in 5 mL of anhydrous tetrahydrofuran is added dropwise, and the cooling bath is removed. The solution is stirred for 1 hr at room temperature and then at reflux for 2 hr. After the solution is cooled to room temperature, it is poured into a 250-mL, round-bottomed flask and the solvents are removed under reduced pressure on a rotary evaporator. The yellow residue is partitioned between 50 mL of ether and 50 mL of saturated sodium chloride. The layers are separated and the aqueous phase is extracted with three 25-mL portions of ether. The combined organic layers are washed with 50 mL of saturated sodium chloride and dried over magnesium sulfate. Magnesium sulfate is removed by filtration, and the excess solvents are then completely removed under reduced pressure on a rotary evaporator. The resulting yellow solid is dried under reduced pressure and transferred to a 100-mL, two-necked, round-bottomed flask that is fitted with a magnetic stirring bar, a nitrogen inlet, and a rubber septum. The flask is charged with 50 mL of anhydrous tetrahydrofuran and flushed thoroughly with dry nitrogen. The resulting solution of the 2-azadiene 6 (Note 20) is cooled to −78°C, and a solution of butyllithium (12.0 mmol) in hexane is added dropwise via syringe. The deeply colored solution is stirred at −78°C for 1 hr, at which time 1.81 g (15.0 mmol) of freshly distilled allyl bromide (Note 21) is added. The cooling bath is removed, and the solution is stirred for 2 hr at room temperature. The reaction is added to 50 mL of 3 N aqueous hydrochloric acid, and the resulting heterogeneous mixture is stirred vigorously for 18 hr at room temperature. After the addition of 50 mL of saturated sodium chloride, the layers are separated, and the aqueous layer is extracted with three 75-mL portions of ether. The combined organic layers are washed with 75-mL portions of saturated aqueous sodium bicarbonate and saturated sodium chloride, and the washings are back washed with a 50-mL portion of ether. The combined organic layers are dried over magnesium sulfate, and the excess solvents are removed under reduced pressure on a rotary evaporator. Distillation of the resulting yellow oil under reduced pressure gives 1.30–1.45 g (75–83%) of pure 2-methyl-2-phenyl-4-pentenal as a colorless liquid, bp 70–73°C (0.1 mm) (Note 22).
2. Notes
1.
Although
N-hydroxymethylphthalimide may be purchased from Aldrich Chemical Company, Inc., it may also be prepared from
phthalimide and
37% aqueous formaldehyde.
8 Material prepared in this way should be dried at room temperature under reduced pressure over
phosphorus pentoxide.
2.
Aqueous
48% hydrobromic acid should be purchased either from Eastman Organic Chemicals or Mallinckrodt (AR) since that obtained from other sources tends to give, for unknown reasons, less satisfactory results.
3.
The temperature should not be allowed to exceed 30°C during the addition.
4.
Removal of
all of the
hydrobromic acid by washing is critical to the success of the next reaction. If the filtrate is not basic after washing with cold
10% aqueous ammonium hydroxide the washing should be continued until the filtrate is basic. Disconnection of the vacuum during each washing is recommended. The final aqueous wash should be no more basic than pH 8–9. Use of a
rubber dam facilitates the filtration and washing.
5.
The proton magnetic resonance spectrum of
2 exhibits the following absorptions (CDCl
3) δ: 5.42 (s, 2 H, CH
2Br), 7.65–7.91 (complex, 4 H, aromatic).
6.
Triethyl phosphite was purchased from Aldrich Chemical Company, Inc.7.
The exothermic reaction usually commences after all of the solid has dissolved. It is important to allow this exothermic reaction, which lasts about 5 min, to run its course without cooling, since premature cooling results in lower yields of impure product which may be difficult to purify.
8.
Use of impure
N-bromomethylphthalimide or incomplete reaction may lead to the formation of a gummy or mushy residue at this stage, and the addition of
petroleum ether (bp
60–68°C) might facilitate crystallization. Alternatively, the crude product may be purified by dissolving it in a minimum volume of anhydrous
ether, addition of
petroleum ether (bp
60–68°C) until the solution turns cloudy and then cooling. Scratching may be necessary to induce crystallization. Several crops of crystals may be collected, but the total yields thus obtained are generally lower than
80%.
9.
The proton magnetic resonance spectrum of product
3 exhibits the following absorptions (CDCl
3) δ: 1.31 (t, 6 H,
J = 7, CH
2CH
3), 3.83–4.26 (complex, 6 H, OCH
2, NCH
2P), 7.60–7.86 (complex, 4 H, aromatic).
10.
Absolute ethanol was purchased from Aaper Alcohol and Chemical Company and used without further purification. Slight heating may be required to effect solution.
11.
Hydrazine, 95%, was purchased from Eastman Organic Chemicals.
12.
Since it may undergo reaction with
benzaldehyde in the subsequent step to give benzaldehyde azine, it is advisable to remove the last traces of
hydrazine by rotating the flask under reduced pressure. The submitters used an
oscillating motor which operates on compressed air or vacuum and is commonly employed with Kugelrohr distilling units. One such motor is available from the Aldrich Chemical Company, Inc.
13.
Diethyl aminomethylphosphonate undergoes decomposition on attempted distillation, but no deterioration of the product was observed if these operations were executed at temperatures not exceeding 30°C.
14.
The proton magnetic resonance spectrum of crude
4 exhibits the following absorptions (CDCl
3) δ: 1.31 (t, 6 H,
J = 7, CH
2CH
3), 2.68 (br s, 2 H, NH
2), 3.00 (d, 2 H,
J = 10, PCH
2N), 3.95–4.27 (complex, 4 H, aromatic).
15.
The proton magnetic resonance spectrum of
5 exhibits the following absorptions (CDCl
3) δ: 1.32 (t, 6 H,
J = 7, CH
2CH
3), 3.85–4.24 (complex, 6 H, CH
2CH
3, PCH
2N), 7.24–7.40 (complex, 3 H,
para, meta Ph CH), 7.64–7.75 (complex, 2 H,
ortho Ph CH), 8.25 (d, 1 H,
J = 5, N=CHPh). This material shows no significant tendency to deteriorate when stored under dry
nitrogen at room temperature.
16.
The apparatus was flame-dried under a flow of dry
nitrogen and then kept under a slight positive pressure of
nitrogen during the reactions by maintaining a slow flow of
nitrogen through a
mercury bubbler.
17.
Tetrahydrofuran was distilled from the
potassium ketyl of benzophenone.
(Caution: See Org. Synth., Coll. Vol. V, 1973, 976 for a warning regarding the purification of tetrahydrofuran.)18.
Butyllithium was prepared by dilution of
90% butyllithium obtained from Lithium Corporation of America with purified
hexane or petroleum ether (bp
60–68°C)
(Note 19). The normality was determined prior to use by titration according to the method of Watson and Eastham.
919.
Hexane was purified by preliminary stirring over concentrated
sulfuric acid and then anhydrous
potassium carbonate followed by distillation. The
hexane thus obtained was then distilled from
sodium wire.
20.
The proton magnetic resonance spectrum of
6 exhibits the following absorptions (CDCl
3) δ: 2.52 (br s, 3 H, C-CH
3), 7.20–7.65 (complex, 9 H, aromatic), 7.86 (m, 2 H, aromatic), 8.33 (br s, 1 H, N=CHPh).
21.
Allyl bromide was purchased from Aldrich Chemical Company, Inc. and distilled from pulverized
calcium hydride and filtered through basic alumina (10 g) immediately prior to use.
22.
The proton magnetic resonance spectrum of
7 exhibits the following absorptions (CDCl
3) δ: 1.32 (s, 3 H, CCH
3), 2.49 (d, 2 H,
J = 7, CH
2CH=CH
2), 4.88 (m, 2 H, CH=CH
2), 5.45 (m, 1 H, CH=CH
2), 7.14 (m, 5 H, aromatic), 9.33 (s, 1 H, CHO).
3. Discussion
The procedure in the present reaction sequence for the preparation of
N-bromomethylphthalimide (
2) is a modification of that reported by Pucher and Johnson.
2 N-Bromomethylphthalimide has also been prepared by treatment of
N-hydroxymethylphthalimide with
phosphorus tribromide.
3 The procedures for the syntheses of the phosphonates
3 and
4 represent modifications of those described by Yamauchi and co-workers.
3,4 Two other routes to
4 have recently been reported by Gross and co-workers.
5 Ratcliffe and Christensen have also recorded the preparation of
diethyl N-benzylideneaminomethylphosphonate (
5) by the condensation of
benzaldehyde with
4 under conditions virtually identical to those detailed herein, but their route to
4 was completely different.
6 The submitters have found that the present method for the syntheses of
2–5 gives reproducibly higher yields and is more reliable and convenient than those alternative procedures.
Diethyl N-benzylideneaminomethylphosphonate (
5) has been previously employed as an intermediate in the synthesis of β-lactam antibiotics
6 and as a reagent for the homologation of aldehydes and ketones via intermediate 2-azadienes.
7 Other derivatives of dialkyl aminomethylphosphonates have also emerged as useful synthetic reagents. For example,
diethyl isocyanomethylphosphonate (8) may be employed for the conversion of aldehydes and ketones to α,β-unsaturated isocyano compounds.
10 Dimethyl diazomethylphosphonate (9)11 has recently been shown to be an effective reagent for the elaboration of aldehydes or alkyl aryl ketones and diaryl ketones to alkynes
12 and for the conversion of dialkyl ketones into aldehydic enol ethers and enamines.
13
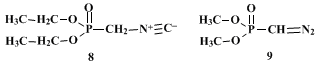
Part D of the present procedure represents a slight modification of a general method for effecting the net replacement of both of the carbon–oxygen bonds of a carbonyl group with an acyl group and a functionalized alkyl appendage; some typical examples of the original procedure are collected in Table I.
14 Moreover, when the electrophile employed for alkylation of the intermediate metalloenamine
15 is properly selected, it is possible to introduce geminal substituents at the carbonyl function that are suitably functionalized for subsequent conversion either to 4,4-disubstituted cyclopentenones (Eq. 1)
14 or 4,4-disubstituted cyclohexenones (Eq. 2).
15 The preparation of
10 represents a formal total synthesis of
α-cuparenone. An annulation related to that depicted in Eq. 2, which was a key step in an efficient synthesis of mesembrine,
16 has also been featured in total syntheses of the Amaryllidaceae alkaloids lycoramine
17 and crinine.
18 Finally, the intermediate metalloenamines may be utilized as the nucleophilic partners in directed aldol reactions (Eq. 3), but it appears to be necessary to
trap the intermediate β-oxidoimines by acylation or alkylation to avoid retroaldolization during the hydrolysis step.
14 Such a process has recently been exploited in the syntheses of the Amaryllidaceae alkaloids pretazettine and haemanthidine.
19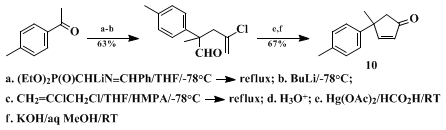
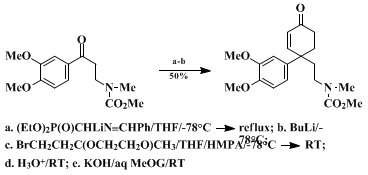
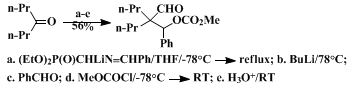
TABLE I
GEMINAL ACYLATION-ALKYLATION OF CARBONYL COMPOUNDS
|
|
|
|
Entry
|
R1
|
R2
|
R3
|
Overall Yield (%)a
|
|
|
1
|
C6H13
|
H
|
Me
|
34
|
|
2
|
c-C6H11
|
H
|
Me
|
43
|
|
3
|
n-Pr
|
n-Pr
|
Me
|
58
|
|
4
|
|
-(CH2)5-
|
CH2CH=CH2
|
51
|
|
5
|
|
-(CH2)2CH(t-Bu)(CH2)2-
|
Me
|
63b
|
|
6
|
|
-CH2C(Me)2CH2C(Me)=CH-
|
Me
|
80
|
|
7
|
Ph
|
Me
|
Me
|
77
|
|
aYields are of distilled products but are not optimized.
|
bAs a ~ 4 : 1 mixture of diastereoisomers.
|
It presently appears that this methodology is well suited for the construction of quaternary
carbon atoms bearing substituted alkyl appendages containing a diverse array of functionality. In large measure this is because metalloenamines, which are the key synthetic intermediates, are highly nucleophilic and generally undergo regioselective reaction at
carbon with a variety of weak and multifunctional electrophiles. Moreover, numerous functional groups may be present on the starting aldehyde or ketone, but one report
20 suggests that carbonyl compounds bearing potential leaving groups on the
carbon adjacent to the carbonyl group may not be good substrates. While a number of individual operations are required, it is frequently possible to execute the entire sequence of reactions in a single flask. In this particular preparation the sequence may also be performed in a single vessel, but purification of product 7 by simple distillation is more difficult because of the presence of lower-boiling impurities.
Appendix
Chemical Abstracts Nomenclature (Collective Index Number);
(Registry Number)
petroleum ether
potassium ketyl of benzophenone
α-cuparenone
ethanol (64-17-5)
potassium carbonate (584-08-7)
sulfuric acid (7664-93-9)
hydrochloric acid (7647-01-0)
Benzene (71-43-2)
ether,
diethyl ether (60-29-7)
formaldehyde (50-00-0)
sodium bicarbonate (144-55-8)
sodium chloride (7647-14-5)
HYDROBROMIC ACID (10035-10-6)
Allyl bromide (106-95-6)
Ethyl bromide (74-96-4)
phosphorus tribromide (7789-60-8)
nitrogen (7727-37-9)
benzaldehyde (100-52-7)
acetone (67-64-1)
Acetophenone (98-86-2)
carbon (7782-42-5)
sodium wire (13966-32-0)
Phthalimide (85-41-6)
ammonium hydroxide (1336-21-6)
hydrazine (302-01-2)
magnesium sulfate (7487-88-9)
butyllithium (109-72-8)
Tetrahydrofuran (109-99-9)
Phthalhydrazide (1445-69-8)
hexane (110-54-3)
Triethyl phosphite (122-52-1)
calcium hydride (7789-78-8)
phosphorus pentoxide (1314-56-3)
2-Methyl-2-phenyl-4-pentenal,
4-Pentenal, 2-methyl-2-phenyl- (24401-39-6)
N-Bromomethylphthalimide (5332-26-3)
Diethyl phthalimidomethylphosphonate (33512-26-4)
Diethyl N-benzylideneaminomethylphosphonate (50917-73-2)
diethyl aminomethylphosphonate (50917-72-1)
diethyl isocyanomethylphosphonate (41003-94-5)
Dimethyl diazomethylphosphonate
N-hydroxymethylphthalimide (118-29-6)
N-bromophthalimide (2439-85-2)
Copyright © 1921-, Organic Syntheses, Inc. All Rights Reserved