Org. Synth. 1988, 66, 95
DOI: 10.15227/orgsyn.066.0095
VINYLATION OF ENOLATES WITH A VINYL CATION EQUIVALENT: trans-3-METHYL-2-VINYLCYCLOHEXANONE
[Cyclohexanone, 2-ethenyl-3-methyl-, trans-]
Submitted by Tony C. T. Chang
1, Myron Rosenblum
2, and Nancy Simms
2.
Checked by Ed Fewkes and Martin F. Semmelhack.
1. Procedure
Caution! Care should be exercised in the preparation of the sodium amalgam since the initial reaction is highly exothermic. This and all subsequent operations should be carried out in a well-ventilated hood. Chloroacetaldehyde diethyl acetal is an irritant and a mutagen. Care should be exercised in its handling.
A. Dicarbonyl(cyclopentadienyl)(ethyl vinyl ether)iron tetrafluoroborate (1). A 500-mL, three-necked flask, with a stopcock at the bottom, is fitted with a nitrogen inlet and a mechanical stirrer with a Teflon paddle. Nitrogen is passed through the flask while it is flame-dried (Note 1) and then 70 mL of mercury is introduced. The mercury is stirred vigorously as 7.2 g (0.31 mol) of sodium metal, cut into small pieces, and added slowly under a strong flow of nitrogen; after which the remaining neck is capped with a rubber septum. The amalgam is allowed to cool to room temperature and 100 mL of tetrahydrofuran is added (Note 2) and (Note 3). While the system is flushed with nitrogen, one septum is removed and 35.4 g (0.1 mol) of dicarbonyl(cyclopentadienyl)diiron [(CO)2CpFe]2 (Note 4) is added at once. Vigorous stirring is continued for 40 min. The mercury is drained through the stopcock, and the deep-yellow-red solution of sodium dicarbonyl(cyclopentadienyl)ferrate, which is ready for use without further purification, is transferred (Note 2) to a 500-mL, round-bottomed flask containing a magnetic stirrer. An additional 50 mL of tetrahydrofuran is used to rinse the amalgam flask.
Chloroacetaldehyde diethyl acetal (31.11 g, 0.20 mol) (Note 5) is added by syringe slowly, since the initial reaction is exothermic. The resulting solution is heated at 50°C with stirring for 2 hr. After the solution is cooled to room temperature, solvent is removed, first with a rotary evaporator and then with an oil pump overnight (Note 6). The residue is taken up in ethyl ether (Note 7), and filtered by suction through a 1½-in. plug of Celite packed in a 250-mL, coarse-porosity, fritted Schlenk tube (Note 8) and (Note 9). The filtrate is collected in a 1-L, round-bottomed flask containing a magnetic stirring bar. The sodium chloride residue is washed several times with fresh ether until the washings are nearly colorless. The filter tube is removed and the flask is capped with a rubber septum and a nitrogen inlet; air is displaced by flushing the flask is capped with nitrogen.
The solution is cooled to −78°C in a dry ice–acetone bath, and 38.19 g (0.23 mol) of tetrafluoroboric acid–diethyl ether complex (Note 10) is added dropwise by syringe over a 30-min period. The solution is allowed to warm to room temperature. The yellow precipitate is filtered off and collected in a 250-mL Schlenk tube, washed with ether, and dried under a stream of nitrogen and finally under reduced pressure (oil pump). The bright-yellow salt 1 weighs 43.2–57.6 g (60–80%) and may be used without further purification (Note 11). It may be stored indefinitely under nitrogen at 0°C without decomposition.
B.
Dicarbonyl(cyclopentadientyl)(trans-3-methyl-2-vinylcyclohexanone)iron tetrafluoroborate (3). Under a
nitrogen atmosphere,
cuprous iodide (Note 12) (24.76 g, 0.13 mol) and
150 mL of ether are placed in a
1-L, three-necked, round-bottomed flask containing a magnetic stirring bar, and cooled to 0°C in an
ice–salt bath. First
172 mL (0.26 mol) of methyllithium in ether (Note 13) is added by syringe, then
12.60 g (0.13 mol) of 2-cyclohexen-1-one (Note 14) is added dropwise by syringe while the mixture is stirred at 0°C. A bright yellow precipitate forms immediately. After 15 min,
200 mL of tetrahydrofuran is added and the mixture is cooled to −78°C in a dry ice–acetone bath. While vigorous stirring and a strong flow of
nitrogen are maintained, one septum is removed and
43.70 g (0.13 mol) of complex salt 1 is added at once. The septum is replaced and
50 mL of fresh tetrahydrofuran is used to wash solid
1 from the neck and sides of the reaction vessel. After 1 hr at −78°C, the mixture is allowed to warm to room temperature while stirring is continued. Stirring is halted to allow insoluble copper salts to settle, leaving a red supernatant liquid. A
250-mL, coarse-frit, Schlenk filter is prepared with a Celite mat, topped with 2 in. of activity-IV neutral alumina
(Note 15), which is further deactivated by washing in the Schlenk tube with
100 mL of diethyl ether. The supernatant solution is transferred to the Schlenk tube by cannula and filtered by suction into a 1-L, round-bottomed flask containing a magnetic stirring bar. The copper salts remaining in the reaction vessel are repeatedly washed with fresh
ether until the filtered washings are nearly colorless. Removal of solvent from the filtrate leaves product
2 as a deep red oil
(Note 16).
The oil is dissolved in 500 mL of diethyl ether under a nitrogen atmosphere, cooled to −78°C in a dry ice–acetone bath and 18 g (0.11 mol) of tetrafluoroboric acid–diethyl ether complex is added dropwise by syringe over a 30-min period, while the bath temperature is maintained at −78°C. The solution is allowed to warm to room temperature, and the powdery yellow solid is isolated by filtration through a 250-mL, coarse-frit, Schlenk filter tube. The product is washed several times with fresh ether and dried under a stream of nitrogen and then under reduced pressure (oil pump). The yield of salt 3 is 31–39.1 g (60–75%). The product may be used without further purification (Note 17) and may be stored under nitrogen of 0°C for several weeks with no observable decomposition (Note 18).
C.
trans-3-Methyl-2-vinylcyclohexanone (4). Compound 3 (31.5 g) and
25 mL of acetonitrile (0.47 mol, 6-fold excess) (Note 19) are placed in a
100-mL round-bottomed flask (Note 20) fitted with a magnetic stirring bar and a
reflux condenser. The mixture is heated to reflux for 2 hr under
nitrogen, cooled to room temperature, and slowly added to
300 mL of diethyl ether. The
acetonitrile complex 5 precipitates as a bright yellow solid, and may be removed by suction filtration
(Note 21).
The filtrate is washed 3 times with distilled water to remove excess acetonitrile and then dried over anhydrous magnesium sulfate. Filtration followed by removal of ether leaves the product 4 as a yellow oil (7.1–7.6 g, 64–70%), which may be further purified by short path or bulb-to-bulb distillation (bp 30°C at 0.1 mm) to a colorless liquid (Note 22) and (Note 23).
2. Notes
1.
All glassware, syringes, and cannulae were routinely flame- or oven-dried and cooled under dry
nitrogen.
2.
In general, transfers of dry solvent or of solutions are made by 2-mm cannulae inserted through rubber septa capping delivery and
receiver vessels. Transfer is made by positive
nitrogen pressure applied through a hypodermic needle, while a second needle in the receiver vessel is employed as a vent. Cannulae are available from Hamilton Company, P.O. Box 10030, Reno, NV 89510.
3.
Tetrahydrofuran is predried over
potassium hydroxide pellets, degassed with
nitrogen, and freshly distilled under
nitrogen from
sodium benzophenone ketyl.
34.
Dicarbonyl(cyclopentadienyl)diiron can be readily prepared on a large scale from
iron pentacarbonyl and
dicyclopentadiene.
4 Alternatively it can be purchased from Alfa Products, Johnson Mathey Co. or from Strem Chemical Company.
5.
Chloroacetaldehyde diethyl acetal was purchased from Aldrich Chemical Company, Inc. This substance is listed as an irritant. Proper care should be exercised when handling it.
6.
It is important to stir the product continually to ensure effective removal of
tetrahydrofuran. If appreciable solvent remains, the vinyl ether complex
1 may not crystallize readily.
7.
Ethyl ether was freshly distilled from
sodium benzophenone ketyl.
8.
Schlenk tubes were purchased from Ace Glass Company, Catalog No. 7761-36.
9.
Filtration is smoothly accomplished by allowing the
sodium chloride to settle and filtering the clear supernatant liquor in small portions, transferring the solution to the filtering tube by cannula. If the Celite should become clogged, the surface may be scraped clean, under a strong stream of
nitrogen, using a
long spatula, in order to increase the filtration rate.
10.
Tetrafluoroboric acid–diethyl ether complex was purchased from Columbia Organics Chemical Company, Inc.11.
The salt may be reprecipitated by dissolution in
methylene chloride containing a small amount of
ethanol, followed by the addition of
ether. The product shows the following spectra: IR (CH
2Cl
2) cm
−1: 2095, 2045, 1545; NMR (CD
3NO
2) δ: 1.40 (t, 3 H, Me), 2.73 (dd, 1 H,
trans-CH=), 3.00 (dd, 1 H,
cis-CH=), 4.37 (q, 2 H, OCH
2), 5.50 (s, 5 H, Cp), 7.92 (dd, 1 H, CHOEt). The checkers realized the higher yield only when the preparation was carried out on one-tenth the scale specified here.
12.
Cuprous iodide was purchased from Fisher Scientific Company, and purified by recrystallization from saturated aqueous
potassium iodide.
5,613.
Methyllithium was purchased from Aldrich Chemical Company, Inc., and standardized by double titration with
benzoic acid in aqueous ethanol and with
allyl chloride.
714.
2-Cyclohexen-1-one was purchased from Aldrich Chemical Company, Inc., and purified by vacuum distillation.
15.
Alumina was purchased from Woelm and brought to activity IV as directed.
16.
As in the preparation of the vinyl ether complex,
1, it is very important to remove
tetrahydrofuran effectively to promote facile crystallization of product. This is most easily done by removing most of the solvent with a rotary evaporator and then stirring the resulting oil under reduced pressure (oil pump) overnight. Mixing the oil with about
50 mL of ether followed by solvent removal under reduced pressure helped to facilitate removal of traces of
tetrahydrofuran. Pure
2 may be obtained as a yellow crystalline solid, which decomposes on heating, by chromatographing the oil on basic alumina (activity IV), eluting with
5% ether in hexane. Compound
2 is characterized by the following spectra: IR (CH
2Cl
2) cm
−1: 2000, 1940, 1700; NMR (CDCl
3) δ: 1.17 (t, 6 H, CH
3), 1.43 (d, 2 H, FeCH
2), 1.65 (dd, 1 H, CHCO), 1.70–2.55 (m, 7 H, CH, CH
2), 3.41 (dq, 2 H, OCH
2), 3.76 (dt, 1 H, CHOEt), 4.87 (s, 5 H, Cp).
17.
Compound
3 may be recrystallized by dissolution in a minimum volume of
nitromethane at 0°C followed by the addition of
diethyl ether. The crystalline material decomposes on heating. It is characterized by the following spectra: IR (CH
2CH
2) cm
−1: 2090, 2050, 1708; NMR (CD
3NO
2) δ: 1.1 (m, 3 H, CH
3), 1.4–2.6 (m, 8 H, CH, CH
2), 3.28 (d, 1 H,
trans-CH
2=), 4.00 (d, 1 H,
cis-CH
2=), 5.0 (m, 1 H, CH=), 5.65 (s, 5 H, Cp).
18.
However, salt
3 is unstable at room temperature as a solid and in solution and rearranges to the isomeric complex in which the iron center is bound to the carbonyl group of the substituted cyclohexanone.
19.
Acetonitrile is freshly distilled under
nitrogen from
calcium hydride.
20.
Because of the ease with which product
4 isomerizes to the conjugated enone in the presence of base, it is imperative that the demetallation and subsequent purification steps be carried out in glassware that is free of basic residues.
21.
An inert atmosphere is not necessary.
22.
The considerable vapor pressure of
4 causes some loss during vacuum distillation. Although the color of the product is improved by distillation, both IR and NMR spectra of the product before and after distillation show little change.
23.
Compound
4 is observed to darken on standing in air at room temperature for prolonged periods. Compound
4 is characterized by the following spectra: IR (neat) cm
−1: 1708; NMR (CDCl
3) δ: 0.99 (d,d, 1 H, CH
3), 1.3–2.8 (m, 8 H, CH, CH
2), 4.98 (dd, 1 H,
trans-CH
2=), 5.2 (dd, 1 H,
cis-CH
2=), 5.76 (m, 1 H, CH=).
3. Discussion
Few reagents are available to the synthetic organic chemist that function as vinyl cation synthons. At present, these include
α-phenylseleneoacetaldehyde,
8 α-silyl aldehydes, and ketones,
9, α,β-epoxysilanes,
10 phenyl vinyl sulfoxide,
11 phenyl ethynyl sulfone,
12,13 phenyl 2-chlorovinyl sulfone,
13 and activated vinyl halides or ethynyl halides.
14 With the exception of the first two, the use of these reagents is confined to reactions with tertiary enolates or organocuprates.
The procedure given here illustrates the use of readily prepared organoiron complex
1 for the vinylation of a secondary enolate. This salt may be prepared on a large scale from readily available starting materials and can be stored at 0°C without decomposition. The closely related
isopropenyl ethyl ether and
cis-propenyl ethyl ether–
iron complexes,
6 and
7, are similarly prepared from
α-bromoacetone15,16 and
α-bromopropionaldehyde diethyl acetal17 and have been used as isopropenylating
16 and
trans-propenylating
18 reagents with enolates. Complex
8, derived from
ethyl 3-bromopyruvate, functions as an α-acrylic ester cation with enolates.
19,20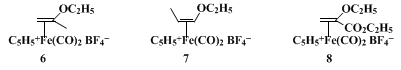
Because of their high reactivity, these complex salts react rapidly and regiospecifically, at low temperature, with a number of carbon and heteroatomic nucleophiles, including thiols, amines, and alcohols.
21 Finally, exposure of the double bond takes place under particularly mild conditions so that isomerization of the β,γ-unsaturated carbonyl system may be avoided. The present scope of reactions with these vinyl cation synthons is summarized in Table I.
TABLE I
VINYLATION OF ENOLATES WITH VINYL CATION EQUIVALENTS
|
|
Enolate
|
Enol Ether Complex
|
Vinylated Product
|
Yield%
|
|
|
|
1
|
|
80
|
|
|
1
|
|
80
|
|
|
1
|
|
80
|
|
|
1
|
|
47a
|
|
|
7
|
|
72
|
|
|
7
|
|
53
|
|
|
7
|
|
91
|
|
|
6
|
|
72
|
|
|
8
|
|
44b 37c
|
|
|
8
|
|
65b
|
|
|
8
|
|
(15)d
|
|
|
|
bBy reduction of the initial condensation product with L-Selectride.
|
cBy reduction of the initial condensation product with sodium borohydride.
|
dBy reduction of the initial condensation product with LiAlH4 at −78°C.
|
22
Appendix
Chemical Abstracts Nomenclature (Collective Index Number);
(Registry Number)
sodium benzophenone ketyl
dicarbonyl(cyclopentadienyl)diiron
Dicarbonyl(cyclopentadientyl)(trans-3-methyl-2-vinylcyclohexanone)iron tetrafluoroborate
acetonitrile complex
α-phenylseleneoacetaldehyde
ethanol (64-17-5)
ether,
ethyl ether,
diethyl ether (60-29-7)
acetonitrile (75-05-8)
iron (7439-89-6)
sodium chloride (7647-14-5)
potassium iodide (7681-11-0)
nitrogen (7727-37-9)
allyl chloride (107-05-1)
Benzoic acid (65-85-0)
mercury (7439-97-6)
potassium hydroxide (1310-58-3)
sodium (13966-32-0)
Nitromethane (75-52-5)
methylene chloride (75-09-2)
cuprous iodide (7681-65-4)
α-bromoacetone (598-31-2)
magnesium sulfate (7487-88-9)
Tetrahydrofuran (109-99-9)
hexane (110-54-3)
Methyllithium (917-54-4)
dicyclopentadiene (77-73-6)
isopropenyl ethyl ether (926-66-9)
chloroacetaldehyde diethyl acetal (621-62-5)
calcium hydride (7789-78-8)
sodium borohydride (16940-66-2)
iron pentacarbonyl
2-cyclohexen-1-one,
cyclohexenone (930-68-7)
ethyl 3-bromopyruvate (70-23-5)
Phenyl vinyl sulfoxide (20451-53-0)
tetrafluoroboric acid (16872-11-0)
Sodium dicarbonyl(cyclopentadienyl)ferrate (12152-20-4)
trans-3-Methyl-2-vinylcyclohexanone,
Cyclohexanone, 2-ethenyl-3-methyl-, trans- (110222-94-1)
phenyl ethynyl sulfone
phenyl 2-chlorovinyl sulfone
α-bromopropionaldehyde diethyl acetal
Dicarbonyl(cyclopentadienyl)(ethyl vinyl ether)iron tetrafluoroborate
cis-propenyl ethyl ether
Copyright © 1921-, Organic Syntheses, Inc. All Rights Reserved