Org. Synth. 1987, 65, 203
DOI: 10.15227/orgsyn.065.0203
(−)-8-PHENYLMENTHOL
[Cyclohexanol, 5-methyl-2-(1-methyl-1-phenylethyl)-, [1R-(1α,2β,5α)]-]
Checked by Lalith R. Jayasinghe and James D. White.
1. Procedure
Caution! Chloroacetyl chloride is a strong lachrymator. N,N-Dimethylaniline is a severe poison. Synthetic work with these substances should be performed in an efficient hood.
A. (2RS,5R)-5-Methyl-2-(1-methyl-1-phenylethyl)cyclohexanone (2). (See (Note 1).)
a. GRIGNARD REAGENT FORMATION AND CONJUGATE ADDITION. In a nitrogen-flushed, 500-mL, two-necked, round-bottomed reaction flask fitted with a reflux condenser carrying a calcium chloride tube, a 250-mL pressure-equalizing dropping funnel, and a Teflon-coated magnetic stirring bar are placed 11.0 g (0.45 mol) of magnesium turnings and 50 mL of diethyl ether (Note 2). To this flask is added 10% of 78.5 g (0.5 mol) of bromobenzene in one portion (Note 3). The reaction mixture is heated to reflux without stirring to start Grignard reagent formation. When the reaction has started (Note 4), the rest of the bromobenzene in 100 mL of diethyl ether is added with stirring at such a rate that gentle reflux is maintained. After the addition is complete, the reaction mixture is heated to reflux for an additional 1 hr. The solution is cooled to room temperature and diethyl ether is added to give a total volume of about 300 mL (Note 5). The reflux condenser and the dropping funnel are replaced by a nitrogen inlet tube and a pierced rubber septum with Teflon tube inlet (Note 6).
In a second
nitrogen-flushed, 500-mL, three-necked, round-bottomed reaction flask with a
mechanical stirrer, a
thermometer, and a
two-way adapter carrying a calcium chloride tube and a rubber septum with a Teflon tube, connected to a reaction flask
(1 in Fig. 1), are placed
4.4 g (31 mmol) of copper(I) bromide (Note 7) and
70 mL of diethyl ether. The ethereal Grignard solution from the reaction flask (1) is added, through the Teflon tube by means of
nitrogen pressure (see
Fig. 1), to this vigorously stirred suspension at −20°C. After the addition is complete, the reaction mixture is stirred at −20°C for ½ hr. The rubber septum is replaced by a
100-mL, pressure-equalizing dropping funnel containing
40.0 g (0.26 mol) of (R)-(+)-pulegone (Note 8) in
50 mL of diethyl ether. This solution is added with stirring at −20°C to the dark-green reaction mixture during ca. 2 hr. After the reaction mixture is kept overnight at −20°C, it is added to
300 mL of vigorously stirred ice-cold 2 N hydrochloric acid. The organic layer is separated and filtered with suction, and the residue on the funnel is washed twice with
20-mL portions of ether. The aqueous layer is saturated with
ammonium chloride and extracted three times with
100-mL portions of ether. The combined organic phases are washed with saturated aqueous
sodium hydrogen carbonate solution and the solvent is evaporated under reduced pressure. The crude oily product (ca.
62.4 g) is used for equilibration without further purification
(Note 9).
Figure 1
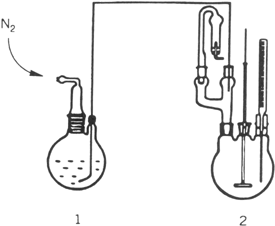
b. E
QUILIBRATION OF K
ETONES (
2). A solution of
62.4 g of crude 2 in
600 mL of ethanol, 80 mL of water, and
70.0 g (1.2 mol) of potassium hydroxide is refluxed for 3 hr. The solution is concentrated on a
rotary evaporator to a volume of about 200 mL, and 500 mL of water is added. This aqueous solution is saturated with
sodium chloride and extracted with four
100-mL portions of ether. The combined organic layers are dried over anhydrous
magnesium sulfate and the solvent is evaporated at reduced pressure. The remaining oily liquid is distilled under reduced pressure at 0.05 mm. Three fractions are collected; the first fraction (boiling range 40–80°C) is discarded. Fraction 2 (boiling range: 80–100°C; 120°C oil-bath temperature) consists mainly of
biphenyl with small amounts of ketone
2 (Note 10). Fraction 3 (boiling range: 100–110°C; 140°C oil-bath temperature) contains the main quantity of ketone
2. Fraction 3 and the decanted liquid of fraction 2 are combined to yield
47.3–54.5 g (
79–91%) of pale-yellow oily
2 (Note 11).
B.
(1RS,2SR,5R)-5-Methyl-2-(1-methyl-1-phenylethyl)cyclohexanol (3/4). In a 500-mL, three-necked, round-bottomed reaction flask fitted with a reflux condenser carrying a calcium chloride tube, a
250-mL pressure-equalizing funnel, and a
mechanical Hershberg stirrer2 are placed
16.0 g (0.70 mol) of sodium and
220 mL of toluene (Note 12). The solution is heated to reflux and maintained at this temperature. By vigorous stirring a fine suspension of
sodium is obtained. To this stirred suspension a solution of
54.5 g (0.24 mol) of equilibrated 2 in
40.8 g (0.68 mol) of 2-propanol (Note 13) is added dropwise at such a rate that controlled refluxing is maintained. After the addition is complete, the reaction mixture is refluxed for an additional 8 hr and then cooled to 0°C. The mixture is diluted with
250 mL of ether (Note 14) and carefully poured into 260 mL of ice water. The organic layer is separated and the aqueous phase is saturated with
sodium chloride and extracted 3 times with
100-mL portions of ether. The combined organic layers are washed with saturated aqueous
sodium chloride solution, dried over anhydrous
magnesium sulfate, filtered, and concentrated by rotary evaporation. Fractional distillation of the concentrate gives
39.0–48.9 g (
70–88%) of pale-yellow
3/4, bp
103–107°C/0.01 mm (126°C oil-bath temperature)
(Note 15).
C.
(1R,2S,5R)-5-Methyl-2-(1-methyl-1-phenylethyl)cyclohexyl chloroacetate 5 (Note 16). A
250-mL, three-necked, round-bottomed reaction flask fitted with a reflux condenser and a calcium chloride tube, a
50-mL pressure-equalizing funnel, a thermometer, and a Teflon-coated magnetic stirring bar is charged with
20.0 g (86 mmol) of 3/4,
10.5 g (86 mmol) of N,N-dimethylaniline, and
30 mL of diethyl ether. This stirred mixture is cooled to 0°C and a solution of
10.5 g (93 mmol) of chloroacetyl chloride in
30 mL of diethyl ether is added at such a rate that this temperature is maintained. After the reaction is stirred at 0°C for an additional hr, the
ice bath is removed and the reaction mixture is allowed to warm to room temperature, during which time
N,N-dimethylaniline hydrochloride precipitates. The reaction is completed by heating to reflux for 3 hr
(Note 17). The solvent is removed under reduced pressure using a rotary evaporator, and the crystalline white residue is dissolved in
60 mL of dichloromethane and 60 mL of water. The phases are separated and the organic phase is washed thoroughly with an equal volume of water; then it is washed until it is acid-free with a saturated aqueous
sodium hydrogen carbonate solution. It is concentrated under reduced pressure to give about
25.0 g of a viscous oil, which crystallizes on addition of
30 mL of 90% aqueous ethanol. The crystals are filtered with suction to yield
18.6–21.8 g (
70–82%) of the chloroacetate as a mixture of diastereomers. Diastereo- and enantiomerically pure chloroacetate
5 is obtained in
48% yield by two fractional crystallizations of the diastereomeric chloroacetates from
ethanol, mp
82–83°C;
[α]D20 + 22.4° (CCl
4,
c 2.29)
(Note 18).
D.
(1R,2S,5R)-5-Methyl-2-(1-methyl-1-phenylethyl)cyclohexanol (3). In a 500-mL round-bottomed reaction flask, fitted with a reflux condenser and a Teflon-coated magnetic stirring bar,
12.8 g (41 mmol) of 5 (48%) is dissolved in a solution of
300 mL of ethanol, 40 mL of water, and
46 g (82 mmol) of potassium hydroxide. This solution is refluxed for 2 hr. The solution is concentrated at reduced pressure to a volume of ca. 50 mL and 200 mL of water and
100 mL of ether are added. After the
ether layer is separated, the aqueous phase is saturated with
sodium chloride and extracted with three
50-mL portions of ether. The combined organic layers are dried over anhydrous
magnesium sulfate, filtered, and the solvent is evaporated. Kugelrohr distillation of the cloudy residual oil yields
8.9–9.2 g (
92–97%) of
3, bp
105–115°C at 0.01 mm;
[α]D20 −26.4° ± 0.1° (
ethanol,
c 1.97)
(Note 19).
2. Notes
1.
Parts A and B of this procedure are based on a communication by E. J. Corey and H. E. Ensley.
3 This protocol has been disclosed previously.
42.
The submitter used
diethyl ether distilled from
sodium wire.
3.
Bromobenzene was purchased from Merck-Schuchardt and was used without further purification.
4.
Sometimes it becomes necessary to add some single crystals of
iodine to start the reaction.
5.
The concentration of the ethereal Grignard solution was estimated to be 1.38
N, as determined by hydrolysis of an aliquot (1 mL taken by syringe) and titration with
0.1 N hydrochloric acid.
6.
The Teflon tube was 3 mm in diameter.
7.
Copper(I) bromide was purchased from Fluka AG, Buchs, Switzerland and was not further purified. In previous runs
copper(I) iodide was used to give comparable yields.
8.
(R)-Pulegone had
[α]D20 + 24.6° (
ethanol,
c 1.92) and was obtained from Haarmann & Reimer, Holzminden. The checkers used
technical-grade (+)-pulegone (82% pulegone content) and obtained
2 in
67–70% isolated yield after equilibration. The submitters thank Haarmann & Reimer, Holzminden, Germany, for generous gifts of pure and
technical-grade (+)-pulegone used in their work.
9.
An epimeric mixture of diasteromeric ratio 55:45 was determined by
13C NMR spectroscopy.
10.
The Grignard coupling product,
biphenyl (mp
70°C, bp
250°C at 760 mm), crystallizes in the condenser and has to be liquified by warming with a heat gun.
11.
The elemental and structural characterization of
2 is as follows. Anal. calcd. for C
16H
22O: C, 83.43; H, 9.63. Found: C, 83.61; H, 9.80; IR (liquid film) cm
−1: 1712 (C=O);
1H NMR (CDCl
3) δ: 0.80–2.78 (m, 8 H), 0.90 (d, 3 H,
J = 6, CH
3CH
cis-
2), 0.96 (d, 3 H,
J = 6, CH
3CH
trans-
2), 1.42 (s, 3 H, CH
3CPh
trans-
2), 1.43 (s, 3 H, CH
3CPh
cis-
2), 1.48 (s, 3 H, CH
3CPh
cis/trans-
2), 7.00–7.44 (m, 5 H, aromatic H);
13C NMR (CDCl
3),
trans-
2 δ: 22.21 (CH
3), 23.52 (CH
3), 26.71 (CH
3), 28.89 (CH
2), 34.51 (CH
2), 36.02 (CH), 38.93 (C
quat), 52.13 (CH
2), 59.23 (CH), 125.37, 125.60, 127.87 and 149.71 (aromatic C), 210.31 (C=O);
cis-
2 δ: 19.03 (CH
3), 23.67 (CH
3), 24.71 (CH
2), 27.23 (CH
3), 31.10 (CH
2), 32.00 (CH), 39.32 (C
quat), 50.07 (CH
2), 59.44 (CH), 125.44, 125.72, 128.65, 149.26 (aromatic C), 211.21 (C=O). An epimeric mixture of diastereomeric ratio 83:17 was determined by
13C NMR spectroscopy. Ketones
2 have
nD20 1.5270–1.5280.
12.
Toluene was distilled from
sodium.
13.
2-Propanol was refluxed with
magnesium methoxide and fractionated.
14.
Without this additional solvent the mixture is quite viscous.
15.
13C NMR-spectroscopy indicated a
3/4 ratio of 87:13. Diastereomers
3/4 can be separated by careful medium-pressure silica gel chromatography (
petroleum ether/ether; 95:5)
5 or by fractional crystallization of the diastereomeric chloroacetates (vide supra). For structural characterization, see
(Note 19).
16.
Part C is based on a report by G. Bergson and co-workers
6 and has been disclosed previously.
4 Compound
5 has also been prepared by using
pyridine/
4-dimethylaminopyridine in
petroleum ether and
chloroacetyl chloride in
benzene.
7 Alternatively,
chloroacetate 5 has been prepared according to a method by Herzog and Scharf.
8 A modification of Bergson's original procedure
6 has been published recently.
917.
The submitters suggest that a revised workup procedure as follows is preferable, but it was not checked. At this point
250 mL of ether and 60 mL of water are added to dissolve the salt. The phases are separated and the procedure as described is followed.
18.
The structural and elemental characterization of
5 is as follows. Anal. calcd. for C
18H
25ClO
2: C, 70.00; H, 8.16. Found: C, 69.81; H, 8.20; IR (KBr) cm
−1: 1185 (COC), 1754 (C=O);
1H NMR (CDCl
3) δ: 0.66–2.24 (m, 8 H), 0.90 (d, 3 H,
J = 6, CH
3CH), 1.22 (s, 3 H, CH
3CPh), 1.33 (s, 3 H, CH
3CPh), 3.04, 3.43 (AB, 2 H,
J = 15, CH
2Cl), 4.91 (dt, 1 H,
J = 10.6, 4, HCO), 7.00–7.40 (m, 5 H, aromatic CH);
13C NMR (CDCl
3) δ: 21.71 (CH
3), 22.72 (CH
3), 26.18 (CH
2), 29.67 (CH
3), 31.22 (CH), 34.37 (CH
2), 39.36 (C
quat), 40.66 (CH
2), 41.43 (CH
2), 50.20 (CH), 75.65 (CH), 124.93, 125.09, 127.79, 151.43 (aromatic C), 166.17 (C=O). The submitters report that
2 may be recovered from the mother liquors enriched in the unwanted diastereomer. The mother liquors were saponified to
3/4 as described in Section D, followed by dichromate oxidation to
2 in
77–81% yield according to a procedure given for
menthone.
10 However, this recovery was not checked.
19.
The optical rotation,
[α]D23 + 26.3° (
ethanol,
c 2.02), for the enantiomer of
3 is reported.
3 The elemental and structural characterization of
3 is as follows. Anal. calcd. for C
16H
24O: C, 82.70; H, 10.41. Found: C, 82.80; H, 10.27; IR (liquid film) cm
−1: 3420, 3570 (OH);
1H NMR (CDCl
3) δ: 0.64–2.06 (m, 9 H), 0.87 (d, 3 H,
J = 6, CH
3CH), 1.29 (s, 3 H, CH
3CPh), 1.42 (s, 3 H, CH
3CPh), 3.48 (dt,
J = 10, 4, HCO), 6.97–7.46 (m, 5 H, aromatic H);
13C NMR (CDCl
3) δ: 21.95 (CH
3), 25.93 (CH
3), 26.62 (CH
2), 27.33 (CH
3), 31.45 (CH), 34.81 (CH
2), 39.87 (C
quat), 45.62 (CH
2), 53.85 (CH), 72.52 (CH), 125.22, 125.52, 127.88, 150.83 (aromatic C). In our hands
3 is a stable compound. In contrast to another report
8 we encountered no difficulties in storing alcohol
3.
3. Discussion
Since its introduction in 1975 by E. J. Corey and H. E. Ensley,
3 8-phenylmenthol has found widespread use as a chiral auxiliary in organic syntheses.
3 5 6 11 12 13 14 15 16 17 18 19 20 21 22 23 24 25 26 27 28 29 30 31 It has proved to be dramatically superior in diastereoface discriminating ability to the commonly used chiral auxiliaries such as
menthol and
borneol.
Starting from
(R)-pulegone we present herein an efficient three-step synthesis furnishing
(−)-8-phenylmenthol as an easily separable 87 : 13-mixture of diastereomers in
55–80% overall isolated yield. Separation of the isomers is achieved either by careful medium-pressure chromatography as reported by Corey and Ensley
3,5 or, less tediously for the preparation of larger amounts, by fractional crystallization of the chloroacetic acid esters
6 and successive saponification as described herein. After submission of this procedure for publication in
Organic Syntheses 1987,
65, 203, a communication
8 was published independently that follows this methodology.
4Recently the isolation of
(−)-8-phenylmenthol via its crystalline phenylcarbamate has been disclosed.
32Since the conversion of
(−)-citronellol to
(S)-pulegone is reported,
33 34 35 the enantiomeric
(+)-8-phenylmenthol likewise may be synthesized. The latter should also be obtainable in a seven-step synthesis starting from
(R)-pulegone (
48% overall yield) as claimed by Corey and co-workers.
5A number of 8-arylmenthols substituted in the aryl moiety have also been synthesized
5,32 and their applicability has been studied in various diastereoselective reactions.
15,32This preparation is referenced from:
Appendix
Chemical Abstracts Nomenclature (Collective Index Number);
(Registry Number)
petroleum ether
Grignard reagent
(−)-8-PHENYLMENTHOL
(−)-citronellol
(1RS,2SR,5R)-5-Methyl-2-(1-methyl-1-phenylethyl)cyclohexanol
ethanol (64-17-5)
hydrochloric acid (7647-01-0)
Benzene (71-43-2)
ether,
diethyl ether (60-29-7)
ammonium chloride (12125-02-9)
sodium hydrogen carbonate (144-55-8)
magnesium turnings (7439-95-4)
sodium chloride (7647-14-5)
nitrogen (7727-37-9)
chloroacetate (79-11-8)
iodine (7553-56-2)
pyridine (110-86-1)
potassium hydroxide (1310-58-3)
toluene (108-88-3)
sodium,
sodium wire (13966-32-0)
2-propanol (67-63-0)
bromobenzene (108-86-1)
Biphenyl (92-52-4)
copper(I) bromide (7787-70-4)
chloroacetyl chloride (79-04-9)
N,N-dimethylaniline (121-69-7)
magnesium methoxide
menthol (15356-60-2)
menthone (1196-31-2)
dichloromethane (75-09-2)
copper(I) iodide (7681-65-4)
magnesium sulfate (7487-88-9)
N,N-dimethylaniline hydrochloride (5882-44-0)
borneol (6627-72-1)
4-dimethylaminopyridine (1122-58-3)
(+)-PULEGONE,
pulegone (89-82-7)
8-phenylmenthol,
(+)-8-phenylmenthol
(1R,2S,5R)-5-Methyl-2-(1-methyl-1-phenylethyl)cyclohexanol,
Cyclohexanol, 5-methyl-2-(1-methyl-1-phenylethyl)-, [1R-(1α,2β,5α)]- (65253-04-5)
(R)-(+)-pulegone,
(R)-Pulegone
(1R,2S,5R)-5-Methyl-2-(1-methyl-1-phenylethyl)cyclohexyl chloroacetate
(S)-pulegone
(2RS,5R)-5-Methyl-2-(1-methyl-1-phenylethyl)cyclohexanone (57707-92-3)
Copyright © 1921-, Organic Syntheses, Inc. All Rights Reserved