Org. Synth. 1989, 67, 60
DOI: 10.15227/orgsyn.067.0060
(−)-SALSOLIDINE
[Isoquinoline, 1,2,3,4-tetrahydro-6,7-dimethoxy-1-methyl-, (S)-]
Submitted by Albert I. Meyers
1, Michael Boes, and Daniel A. Dickman.
Checked by Melinda Gugelchuk and Leo A. Paquette.
1. Procedure
Caution! tert-Butylithium is extremely pyrophoric and must not be allowed to come into contact with the atmosphere. This reagent should only be handled by individuals trained in its proper and safe use. It is recommended that transfers be carried out by using a 20-mL or smaller glass syringe filled to no more than 2/3 capacity, or by cannula. For a discussion of procedures for handling air-sensitive reagents, see Aldrich Technical Bulletin AL-134. [Note added August 2009]
A. 6,7-Dimethoxy-1,2,3,4-tetrahydro-2-[(1-tert-butoxy-3-methyl)-2-butyliminomethyl]isoquinoline (2). In a 250-mL, round-bottomed flask 10.0 g (51.7 mmol) of 6,7-dimethoxy-1,2,3,4-tetrahydroisoquinoline 1 (Note 1) is combined with 11.5 g (53.7 mmol) of (S)-N,N-dimethyl-N'-(1-tert-butoxy-3-methyl)-2-butylformamidine (Note 2), 50 mL of dry toluene, and 50 mg of (+)-camphorsulfonic acid (Note 3). The mixture is heated to reflux for 24 hr and allowed to cool to room temperature. Approximately 30 mL of toluene is removed by rotary evaporation and the residual solution is heated at reflux for an additional 2 days. After the reaction mixture is cooled, it is diluted with 50 mL of dichloromethane and washed with 50 mL of 1 N sodium hydroxide and 100 mL of brine and the organic layer is dried over anhydrous potassium carbonate, filtered, and concentrated by rotary evaporation. The residue is distilled (Kugelrohr 0.1 mm, 170°C bath temp.) to give 18.0 g (96%) of 2 as a pale-yellow oil (Note 4).
B. (S)-6,7-Dimethoxy-1-methyl-1,2,3,4-tetrahydroisoquinoline, (−)-salsolidine (3). A 500-mL, three-necked flask, containing a magnetic stirring bar, is equipped with a three-way stopcock, a low-temperature thermometer, and a rubber septum. The flask is charged with 15.0 g (41.4 mmol) of formamidine 2, filled with argon, and kept under a pressure of ca. 100 mm against the atmosphere (Note 5). Through the septum, via a syringe, is added 300 mL of dry tetrahydrofuran (Note 6) and the solution is cooled to −75°C in a dry ice–acetone bath. A tert-butyllithium solution (21 mL of a 2.4 M solution, (Note 7)), is added dropwise within 5 min through the septum. After the deep-red solution is stirred at −75°C for 45 min, is cooled to −100°C in a liquid nitrogen–methanol bath and, after 15 min at −100°C, 3 mL of freshly distilled iodomethane (Note 8) dissolved in 10 mL of dry tetrahydrofuran is added by syringe at such a rate that the temperature of the reaction mixture does not rise above −90°C. Stirring is continued for 3 hr and the solution is poured into a 1-L separatory funnel containing 50 mL of water. This is extracted twice with 100 mL of dichloromethane and the combined organic layers are washed with 100 mL of brine, dried over potassium carbonate, and filtered. Removal of the solvent on a rotary evaporator gives a cloudy yellow oil, which is dissolved in 100 mL of 60% ethanol. To this solution is added 4.5–5.0 mL of hydrazine (Note 9) followed by 3.0 mL of glacial acetic acid (pH 8–9). The mixture is stirred overnight at ambient temperature and diluted with 50 mL of water. It is extracted twice with 100 mL of dichloromethane, and the combined organic extracts are washed with 50 mL of water, dried (potassium carbonate), filtered, and concentrated at ambient temperature under aspirator pressure. The residue, which consists of valinol tert-butyl ether and salsolidine, is distilled bulb-to-bulb under aspirator pressure at 105°C (pot temperature). This removes the valinol tert-butyl ether (Note 10), leaving crude salsolidine as the pot residue. The residue is dissolved in 100 mL of ether and washed twice with 35-mL portions of ice water–3 N hydrochloric acid (1:4). The ether layer is discarded and the acidic aqueous layer is neutralized with cold (0–5°C) aqueous 25% sodium hydroxide until it is alkaline to pH paper. The creamy mixture is immediately extracted twice with 50 mL of dichloromethane and the organic layers are drawn off, combined, and dried over anhydrous potassium carbonate. After the drying agent is removed by filtration, it is washed twice with 5 mL of dichloromethane. The filtrate and wash are concentrated by rotoevaporation, leaving a yellow oil. Distillation (Kugelrohr) at a pot temperature of 120–125°C (0.01 mm) gives 5.1–5.3 g (60–63%) of (S)-6,7-dimethoxy-1-methyl-1,2,3,4-tetrahydroisoquinoline (salsolidine) 3 as a pale-yellow oil, which crystallizes on standing (Note 11), mp 47–49°C, (Note 12) and (Note 13).
2. Notes
1.
The
tetrahydroisoquinoline hydrochloride was purchased from Aldrich Chemical Company, Inc. and treated with aqueous
5% sodium hydroxide, extracted with
dichloromethane, dried over
sodium sulfate, and distilled, bulb-to-bulb at 0.01 mm (58–60°C, bath temp.) to give a colorless oil. The compound solidifies to give an amorphous solid: mp
83.0–84.5°C; 1H NMR (CDCl
3, 300 MHz) δ: 2.30 (br s, 1 H), 2.69, (t, 2 H,
J = 6), 3.12 (t, 2 H,
J = 6), 3.80 (s, 3 H), 3.82 (s, 3 H), 3.95 (s, 2 H), 6.50 (s, 1 H), 6.58 (s, 1 H).
3.
(+)-10-Camphorsulfonic acid was purchased from Aldrich Chemical Company, Inc.4.
The physical properties are as follows:
1H NMR (CDCl
3, 300 MHz) δ: 0.87 (d, 3 H,
J = 6.8), 0.87 (d, 3 H,
J = 6.7), 1.14 (s, 9 H), 1.83 (hept, 1 H,
J = 7), 2.68–2.81 (m, 3 H), 3.20 (dd, 1 H,
J1 = 8.8,
J2 = 7.1), 3.43–3.56 (m, 3 H), 3.83 (s, 3 H), 3.85 (s, 3 H), 4.40 (AB, 1 H,
J = 17), 4.43 (AB, 1 H,
J = 17), 6.60 (s, 1 H), 6.63 (s, 1 H), 7.40 (s, 1 H);
[α]D25 −30.3° (CHCl
3,
c 2.7).
5.
The flask was filled with
argon by evacuating and pressurizing several times through the three-way stopcock.
6.
Tetrahydrofuran was distilled from
sodium wire and
benzophenone.
7.
tert-Butyllithium solution in pentane was purchased form Alfa Products, Morton/Thiokol, Inc.8.
Iodomethane was purchased from Aldrich Chemical Company, Inc.9.
Anhydrous hydrazine was purchased from Aldrich Chemical Company, Inc.10.
The spectral properties are as follows:
1H NMR (CDCl
3) δ: 0.92 (d, 6 H,
J = 6), 1.19 (s, 9 H), 1.61 (m, 1 H), 2.64 (m, 1 H), 3.12 (overlapping doublets, 1 H,
J = 8,
J = 9), 3.40 (dd, 1 H,
J = 8,
J = 9).
11.
The checkers did not observe crystallization at this point when the reaction was run on a much smaller scale (8 mmol). Two runs, each starting with
3.05 g of
2, gave rise to
0.88 g (
51%) and
0.83 g (
46%) of purified
(−)-salsolidine.
12.
The physical properties are as follows:
1H NMR (300 MHz, CDCl
3) δ: 1.41 (d, 3 H,
J = 6.6), 1.78 (br s, 1 H), 2.65 (m, 1 H), 2.77 (m, 1 H), 3.01 (m, 1 H), 3.25 (m, 1 H), 3.822 (s, 3 H), 3.828 (s, 3 H), 3.89 (m, 1 H), 6.61 (s, 1 H), 6.55 (s, 1 H);
[α]D22 −53.9 to −54.0° (C
2H
5OH,
c 3.8) (lit.
2 −59.5 ± 0.5°); (95–98% ee by Pirkle HPLC analysis,
(Note 13)).
13.
Enantiomeric purity determination was performed as follows. The
(−)-salsolidine 3 (100 mg) was dissolved in
1 mL of dichloromethane and
0.05 mL of triethylamine. To this solution was added
150 mg of 1-naphthoyl chloride (Aldrich Chemical Company, Inc.) and the reaction mixture was stirred for 0.5 hr at room temperature, then poured into
5 mL of aqueous 20% sodium hydroxide solution. Extraction with
10 mL of dichloromethane was followed by separation of the organic layer, which was dried over
potassium carbonate. The solvent was removed under reduced pressure and the crude
naphthamide was purified by column chromatography on
silica gel (
ethyl acetate–hexane–dichloromethane, 1:4:5). Enantiomeric analysis was performed using a Waters Associates Model 440 high-pressure liquid chromatograph equipped with a
Pirkle Covalent Phenylglycine Column (Baker Bond Chiral HPLC Column, DNBPG, J. T. Baker, Phillipsburg, NJ). The detector used was UV at 254 mm and the solvent for elution was
10% 2-propanol–hexane at 5 mL/min with a back pressure of 3500 psi. The peaks were baseline separated and electronically integrated.
3. Discussion
This method of asymmetric alkylation has been performed in a number of other systems with equally good enantioselectivity. Tetrahydroisoquinolines have been alkylated
3 (Eq. 1) with various alkyl halides to give 1-substituted tetrahydroisoquinolines in 50–70% overall yields and with excellent ee values. Several naturally occurring isoquinoline alkaloids have also been prepared (compounds A–C) in 95–98.5% ee.
4 A number of chiral auxiliaries other than the valine-based
tert-butyl ether also have been examined and gave 80–99% ee values after alkylation.
5 However, the authors consider the chiral auxiliary used in the present procedure to be superior to the others.
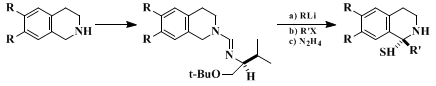
In addition to isoquinolines, β-carbolines have been used to afford indole alkaloids, both natural and unnatural (Scheme 1), in high enantiomeric excess.
6 The indole nitrogen is protected as the methoxymethyl ether and later removed to provide the unsubstituted indole. In the absence of indole–nitrogen protection, the potassium salt is satisfactory but results in low asymmetric induction. However, if racemic products are desired,
N-tert-butylformamidines can be used,
7 and smooth alkylation of the α-protons is achieved, thus obviating the need for protection of the indole nitrogen.
TABLE I
|
|
R,R
|
R'X
|
% ee
|
|
|
H
|
MeI
|
99
|
|
H
|
n-BuI
|
96
|
|
H
|
Allyl Br
|
96
|
|
H
|
PhCH2Cl
|
98
|
|
MeO, MeO
|
3,4-(MeO)2PhCH2Br
|
99
|
|
MeO, MeO
|
3,4-(MeO)2PhCH2CH2I
|
95
|
|
Asymmetric alkylations also are feasible, leading to the chiral
dihydropyrrole (D) and the
tetrahydropiperidine systems (E).
8 When the saturated analogs were employed (
pyrrolidine and
piperidine), no metalation could be effected in the presence of the chiral auxiliary, although metalation–alkylation proceeded normally when the
N-tert-butylformamidines were employed.
This preparation is referenced from:
Appendix
Chemical Abstracts Nomenclature (Collective Index Number);
(Registry Number)
brine
(−)-SALSOLIDINE
6,7-dimethoxy-1,2,3,4-tetrahydroisoquinoline 1
(S)-N,N-dimethyl-N'-(1-tert-butoxy-3-methyl)-2-butylformamidine
N-tert-butylformamidines
ethanol (64-17-5)
potassium carbonate (584-08-7)
hydrochloric acid (7647-01-0)
acetic acid (64-19-7)
ethyl acetate (141-78-6)
ether (60-29-7)
sodium hydroxide (1310-73-2)
sodium sulfate (7757-82-6)
toluene (108-88-3)
Benzophenone (119-61-9)
sodium (13966-32-0)
2-propanol (67-63-0)
piperidine,
tetrahydropiperidine (110-89-4)
iodomethane (74-88-4)
Pentane (109-66-0)
hydrazine (302-01-2)
dichloromethane (75-09-2)
1-naphthoyl chloride (879-18-5)
naphthamide (2243-81-4)
Tetrahydrofuran (109-99-9)
pyrrolidine (123-75-1)
hexane (110-54-3)
triethylamine (121-44-8)
argon (7440-37-1)
Formamidine
tert-Butyllithium (594-19-4)
tetrahydroisoquinoline hydrochloride (14099-81-1)
dihydropyrrole
(+)-camphorsulfonic acid,
(+)-10-Camphorsulfonic acid (3144-16-9)
Isoquinoline, 1,2,3,4-tetrahydro-6,7-dimethoxy-1-methyl-, (S)-,
(S)-6,7-Dimethoxy-1-methyl-1,2,3,4-tetrahydroisoquinoline (493-48-1)
6,7-Dimethoxy-1,2,3,4-tetrahydro-2-[(1-tert-butoxy-3-methyl)-2-butyliminomethyl]isoquinoline (90482-03-4)
valinol tert-butyl ether
Salsolidine
Copyright © 1921-, Organic Syntheses, Inc. All Rights Reserved