Org. Synth. 1999, 76, 86
DOI: 10.15227/orgsyn.076.0086
(2S,3S)-(+)-(3-PHENYLCYCLOPROPYL)METHANOL
[
Cyclopropanemethanol, 2-phenyl-, (1S-trans)-
]
Submitted by André B. Charette and Hélène Lebel
1
.
Checked by Kevin Minbiole, Patrick Verhoest, and Amos B. Smith, III.
1. Procedure
A.
Butylmagnesium bromide
. To a
500-mL, three-necked, round-bottomed flask equipped with an
egg-shaped magnetic stirrer,
125-mL pressure-equalizing addition funnel,
reflux condenser and a
glass stopper
(Note 1), is added
17.0 g (0.70 mol) of magnesium turnings
(Note 2). Stirring is started, and the system is flame-dried for 2 min. The flask is cooled to room temperature under a flow of
argon, and
30 mL of ether
(Note 3) is introduced to cover the
magnesium. A solution of
24 mL (0.22 mol) of bromobutane
(Note 4) in
70 mL of ether
is placed in the pressure-equalizing addition funnel. Then,
1 mL (0.01 mol) of bromobutane
is added to the suspension of
magnesium in
ether. The mixture is heated gently to initiate the reaction
(Note 5). When the reaction has started, the solution of bromide in
ether is added dropwise at a rate sufficient to maintain a gentle reflux
(Note 6). After completion of the addition, the funnel is rinsed with
5 mL of ether
. The gray solution is stirred for 15 min and then transferred to a dry flask under argon via
cannula. The Grignard reagent is titrated with a solution of
isopropyl alcohol
in
benzene using
1,10-phenanthroline
as the indicator
(Note 7).
2 A 1.90-2.10 M solution of Grignard reagent is obtained.
B.
Butylboronic acid
. To a 1-L, one-necked, round-bottomed flask equipped with an egg-shaped magnetic stirrer (Note 1) and an internal thermocouple probe (Note 11) is added
220 mL of ether
(Note 3), followed by
10 mL (89.2 mmol) of trimethyl borate
(Note 12). The clear solution is cooled to −75°C (internal temperature) and stirred vigorously, then
45 mL (87.8 mmol) of a 1.95 M solution of butylmagnesium bromide
in ether
(Note 13) is added dropwise via cannula at such a rate that the internal temperature does not exceed −65°C (Note 14). After the addition is complete, the resulting white slurry is stirred for an additional 2 hr at −75°C under argon. The cooling bath is removed, and the reaction mixture is allowed to warm to room temperature (Note 15). Hydrolysis is carried out by the dropwise addition of
100 mL of an aqueous 10% solution of hydrochloric acid
. The white precipitate is dissolved, and the resulting clear biphasic mixture is stirred for 15 min, at which time the two layers are separated. The aqueous layer is extracted with
ether (2 × 50 mL), and the combined extracts are dried over
magnesium sulfate
. After concentration of the ethereal solution under reduced pressure, the residual white solid is purified by recrystallization as follows: After dissolution in hot water (25 mL), the resulting biphasic solution is cooled to 0°C to induce recrystallization of the boronic acid. The solid is collected on a Büchner funnel, washed with
50 mL of hexanes and placed under vacuum (0.2 mm) for 60 min (Note 16) and (Note 17). Between 5.0-6.5 g (55-72% yield) of the boronic acid is produced as a white solid (Note 18).
C.
[(2-)-N,O,O'[2,2'-Iminobis[ethanolato]]]-2-butylboron,
1. A 250-mL, one-necked, round-bottomed flask equipped with an egg-shaped magnetic stirrer (Note 1) is charged with
5.15 g (50.5 mmol) of butylboronic acid
and
5.31 g (50.5 mmol) of diethanolamine
(Note 19).
Ether, 100 mL
(Note 3) and
50 mL of dichloromethane
(Note 20) are added, followed by about
10 g of molecular sieves 3Å
(Note 21). The resulting heterogeneous solution (Note 22) is stirred for 2 hr under argon. The solid is triturated with
dichloromethane (50 mL), filtered through a medium fritted disk funnel and washed with
dichloromethane (2 × 50 mL). The filtrate is concentrated under reduced pressure to produce the crude desired complex. The diethanolamine complex is purified by recrystallization as follows: the white solid is dissolved in hot
dichloromethane (20 mL), then
ether (50 mL) is added to induce recrystallization of the complex. The mixture is cooled to 0°C and the solid is collected on a Büchner funnel and washed with
ether (2 × 30 mL). The product is dried under reduced pressure (0.2 mm) to afford 7.70 g (89%) of the title compound as a white crystalline solid (Note 23).
D.
(4R-trans)-2-Butyl-N,N,N',N'-tetramethyl[1,3,2]dioxaborolane-4,5-dicarboxamide
3. A 500-mL, one-necked, round-bottomed flask equipped with an egg-shaped magnetic stirrer, under argon is charged with
7.70 g (45.0 mmol) of the butylboronate diethanolamine complex and
11.9 g (58.3 mmol) of (R,R)-(+)-N,N,N',N'-tetramethyltartaric acid diamide
(Note 24). The solids dissolve upon the addition of
225 mL of dichloromethane
.
Brine (70 mL) is added, and the resulting biphasic solution is stirred for 30 min under argon. The two layers are separated, and the aqueous layer is extracted with
dichloromethane (50 mL). The combined organic layers are washed with
brine (50 mL), dried over magnesium sulfate and filtered. The filtrate is concentrated under reduced pressure and dried under reduced pressure (0.2 mm) to give 11.3 g (93%) of the title compound as a pale yellow oil (Note 25).
E.
(2S,3S)-(+)-(3-Phenylcyclopropyl)methanol
. A 250-mL, one-necked, round-bottomed flask equipped with an egg-shaped magnetic stirrer (Note 26) and an internal thermocouple probe (Note 11), is charged with
45 mL of dichloromethane
(Note 20) and
1.60 mL (14.9 mmol) of 1,2-dimethoxyethane (DME)
(Note 27). The solution is cooled to −10°C (internal temperature) with an acetone/ice bath, and
1.50 mL (14.9 mmol) of diethylzinc
is added (Note 28). To this stirred solution is added
2.40 mL (29.8 mmol) of diiodomethane
(Note 29) over a 15-20 min period while maintaining the internal temperature between −8°C and −12°C. After the addition is complete, the resulting clear solution is stirred for 10 min at −10°C. A solution of
2.41 g (8.94 mmol) of the dioxaborolane
ligand in
10 mL of dichloromethane
is added via cannula under argon over a 5-6 min period while maintaining the internal temperature below −5°C. A solution of
1.00 g (7.45 mmol) of cinnamyl alcohol
(Note 30) in
10 mL of dichloromethane
is immediately added via cannula under argon over a 5-6 min period while maintaining the internal temperature under −5°C. The cooling bath is removed, and the reaction mixture is allowed to warm to room temperature and stirred for 8 hr at that temperature (Note 31).
Workup.
Method A. The reaction is quenched with aqueous saturated
ammonium chloride (10 mL) and aqueous
10%
hydrochloric acid (40 mL). The mixture is then diluted with
ether (60 mL) and transferred to a separatory funnel. The reaction flask is rinsed with
ether (15 mL), and aqueous
10%
hydrochloric acid (10 mL) and both solutions are transferred to the separatory funnel. The two layers are separated, and the aqueous layer is washed with
ether (20 mL). The combined organic layers are transferred to an Erlenmeyer flask, and a solution containing
60 mL of aqueous 2 N sodium hydroxide
and
10 mL of aqueous 30%
hydrogen peroxide
is added in one portion (Note 32). The resulting biphasic solution is stirred vigorously for 5 min. The two layers are separated and the organic layer is washed successively with aqueous
10%
hydrochloric acid (50 mL), aqueous saturated
sodium sulfite (50 mL), aqueous saturated
sodium bicarbonate (50 mL), and
brine (50 mL). The organic layer is dried over magnesium sulfate and filtered, and the filtrate is concentrated under reduced pressure. The crude product is left under reduced pressure (0.2 mm) overnight (12-16 hr) to remove the butanol produced in this oxidative work-up. The product is purified by a Kugelrohr distillation (90°C, 0.8 mm) to afford 1.05 g (95%) of
(2S,3S)-(+)-(3-phenylcyclopropyl)methanol
as a colorless oil (Note 33) and (Note 34).
Workup. Method B [with recovery of (R,R)-(+)-N,N,N',N'-tetramethyltartaric acid diamide]. The mixture is quenched with aqueous saturated
ammonium chloride (80 mL), and the resulting biphasic mixture is stirred for 5 min. The two clear layers are separated, and the aqueous layer is washed with
dichloromethane (20 mL)
(Note 35). The combined organic layers are dried over magnesium sulfate and filtered, and the filtrate is concentrated under reduced pressure. The residual oil is dissolved in
ether (75 mL) and water (50 mL). The resulting biphasic mixture is stirred for 1 hr. The layers are separated, and the aqueous layer is washed with
ether (20 mL). This aqueous layer is kept for tetramethyltartaric acid diamide recovery (see below). The combined organic layers are treated with
60 mL of aqueous 2 N sodium hydroxide
and
10 mL of aqueous 30%
hydrogen peroxide
(Note 32). The resulting biphasic mixture is stirred for 5 min. The two layers are separated and the organic layer is washed successively with aqueous
10%
hydrochloric acid (50 mL), saturated aqueous
sodium sulfite (50 mL), saturated aqueous
sodium bicarbonate (50 mL), and
brine (50 mL). The organic layer is dried over magnesium sulfate and filtered, and the filtrate is concentrated under reduced pressure. The crude product is left under reduced pressure (0.2 mm) overnight (12-16 hr) to remove butanol produced in this oxidative work-up. The product is purified by a Kugelrohr distillation (90°C, 0.8 mm) to afford 1.02 g (93%) of (2S,3S)-(+)-(3-phenylcyclopropyl)methanol as a colorless oil.
Recovery of (R,R)-(+)-N,N,N',N'-tetramethyltartaric acid diamide. The aqueous layer from the above extraction is concentrated under reduced pressure, and the crude product is recrystallized by an initial dissolution in hot
dichloromethane (5 mL) followed by the addition of
ethyl acetate (10 mL) to afford between 600 mg to 750 mg (33-41% yield) of the (R,R)-(+)-N,N,N',N'-tetramethyltartaric acid diamide
(Note 36) and (Note 37).
2. Notes
1.
All glassware was dried in an
oven (110°C) and after assembly was allowed to cool under an atmosphere of
argon.
2.
Magnesium turnings were purchased from Sigma-Aldrich Fine Chemicals Company Inc.
and were used without further purification.
3.
Ether was freshly distilled from
sodium/benzophenone.
4.
Bromobutane was purchased from Fisher Scientific Company
and was freshly distilled from
phosphorus pentoxide (P
2O
5) (bp
100-104°C).
5.
The formation of a gray cloudy suspension indicates that the reaction has started. Furthermore, the reaction is sufficiently exothermic to induce the ether to reflux even when the reaction flask is not heated. If the reaction does not start within 2 to 3 min, repeat the heating procedure.
6.
Between 1.5 hr and 2 hr are needed for addition.
7.
A dried
10-mL, one-necked, round-bottomed flask is charged with 1 mL of Grignard, some drops of THF
(Note 8) and a crystal of
1,10-phenanthroline
(Note 9). The slightly pink solution is titrated with a 0.5 M solution of
isopropyl alcohol in
benzene
(Note 10). Between 3.8 and 4.2 mL (±0.2 mL) is obtained to give a clear colorless solution (three titrations).
8.
THF was freshly distilled from
sodium/benzophenone.
9.
1,10-Phenanthroline was purchased from Sigma-Aldrich Fine Chemicals Company Inc.
and was used without further purification.
10.
Isopropyl alcohol was freshly distilled from calcium hydride (CaH
2) and
benzene was freshly distilled from
sodium.
11.
A
Barnant 100, Type T Thermo-Couple Thermometer was used to monitor the internal temperature of the reaction solution.
12.
Anhydrous
trimethyl borate (with <5% of methanol) was purchased from Sigma-Aldrich Fine Chemicals Company Inc.
and was used without further purification. Alternatively, a non anhydrous reagent can be dried by distillation from
calcium hydride (bp
68-69°C).
13.
Commercially available
(Sigma-Aldrich Fine Chemicals Company Inc.), butylmagnesium chloride
, 2.0 M in
ether, can be used and a similar yield is observed.
14.
Between 20 and 30 min are needed for the addition.
15.
Between 1 hr and 1.5 hr are needed.
16.
The amount of the
boroxine significantly increases if the solid is left under reduced pressure for a longer period of time. The boroxine is always a contaminant of the boronic acid (see discussion).
17.
Sometimes a second recrystallization is needed to obtain pure
boronic acid by removing by-products resulting from autooxidation.
18.
The physical properties are as follows: mp
95-97°C;
1H NMR (400 MHz, DMSO) δ: 0.56 (t, 2 H, J = 7.6), 0.83 (t, 3 H, J = 7.2), 1.31-1.19 (m, 4 H), 7.34 (br s, 2 H)
;
13C NMR (100 MHz, DMSO) δ: 13.9, 15.3 (br), 25.1, 26.5
;
11B NMR (128.4 MHz, DMSO) δ: 32.7
.
19.
Diethanolamine was purchased from Fisher Scientific Company
and was used without further purification.
20.
Dichloromethane was freshly distilled from CaH
2.
21.
Molecular sieves, 3Å, powder, average particle size 3-5 μ were purchased from Sigma-Aldrich Fine Chemicals Company Inc.
and dried under vacuum at 250°C for 24 hr, before using.
22.
A few minutes after the addition of the molecular sieves, a white precipitate forms and sometimes maintaining stirring becomes difficult.
23.
The physical properties are as follows: mp
145-148°C;
1H NMR (400 MHz, CDCl
3) δ: 0.44-0.48 (m, 2 H), 0.88 (t, 3 H, J = 7.1), 1.21-1.37 (m, 4 H), 2.79 (br s, 2 H), 3.26 (br s, 2 H), 3.88 (br s, 2 H), 3.98 (br s, 2 H), 4.80-4.98 (m, 1 H)
;
13C NMR (100 MHz, CDCl
3) δ: 14.1, 18.4 (br), 26.5, 28.1, 51.4, 62.5
;
11B NMR (128.4 MHz, CDCl
3) δ: 13.1, 32.7
. Anal. Calcd for C
8H
18BNO
2: C, 56.18; H, 10.61; N, 8.19. Found: C, 56.15; H, 10.86; N, 8.07.
24.
(R,R)-(+)-N,N,N',N'-Tetramethyltartaric acid diamide was prepared from
diethyl tartrate and
dimethylamine and was freshly recrystallized with
methanol and
ethyl acetate.
3 The physical properties are as follows: mp
186-187°C [lit.
2 189-190°C];
1H NMR (400 MHz, CDCl
3) δ: 3.01 (s, 6 H), 3.13 (s, 6 H), 4.21 (br s, 2 H), 4.65 (s, 2 H)
;
13C NMR (100 MHz, CDCl
3) δ: 36.1, 36.9, 69.8, 170.8
;
[α]20
D
+43° (EtOH,
c 2.03) [lit.
2
[α]20
D
+43° (EtOH,
c 3.0)]. This product is also commercially available from Sigma-Aldrich Fine Chemicals Company Inc.
25.
The physical properties are as follows:
1H NMR (400 MHz, CDCl
3) δ: 0.85 (t, 2 H, J = 7.7); 0.87 (t, 3 H, J = 7.2), 1.29-1.41 (m, 4 H), 2.98 (s, 6 H), 3.20 (s, 6 H), 5.53 (s, 2 H)
;
13C NMR (100 MHz, CDCl
3) δ: 9.9 (br), 13.6, 25.0, 25.7, 35.7, 36.9, 75.6, 168.23
;
11B NMR (128.4 MHz, CDCl
3) δ: 34.2
;
[α]20
D
−104.4° (CHCl
3,
c 1.70). HRMS Calcd for C
12H
23BN
2O
4: 270.1751. Found: 270.1746. Anal. Calcd for C
12H
23BN
2O
4: C, 53.35; H, 8.58; N, 10.37. Found: C, 53.67; H, 9.07; N, 10.21.
26.
All glassware was flame dried, then cooled under a flow of dry
argon.
27.
DME was freshly distilled from
sodium/benzophenone.
28.
Diethylzinc is a moisture sensitive and pyrophoric liquid and must be manipulated in an inert atmosphere with gas-tight syringes. Neat
diethylzinc was purchased from Akzo Nobel Chemicals Company Inc.
and was used without further purification.
29.
Diiodomethane was purchased from Acros-Fisher Scientific Company
and used without further purification. If necessary,
diiodomethane can be purified if it shows any signs of slight decomposition (orange or red color develops over time):
diiodomethane is washed with aqueous saturated
sodium sulfite, dried over
sodium sulfate (Na
2SO
4), and distilled from
copper (40°C, 1.0 mm). The pale yellow liquid is collected on
copper.
30.
Cinnamyl alcohol was purchased from Sigma-Aldrich Fine Chemicals Company Inc.
and was freshly purified by Kugelrohr distillation: a first fraction boiling at <70°C (1.0 mm) was discarded, and the alcohol was collected as a white solid at 80°C (1.0 mm).
31.
A similar yield is obtained when the submitters stirred this mixture for 14 hr. No noticeable decomposition and side reactions are observed after slightly longer periods of time.
32.
Hydrogen peroxide was purchased from ACP Chemicals Company Inc.
and used without further purification.
33.
Alternatively, the product can be purified by flash chromatography on silica gel (78.5 g, 4 cm × 16 cm) using
30%
ethyl acetate
in hexanes as the mobile phase (800 ml) to afford
1.06 g (
96%) of the title compound.
34.
The physical properties are as follows: bp
90°C, 0.8 mm; IR (film) cm
−1: 3350, 3050, 3000, 2950, 2900, 1600, 1500, 1450, 1100, 1050, 1000, 750, 700
;
1H NMR (400 MHz, CDCl
3) δ: 0.92-1.01 (m, 2 H), 1.43-1.51 (m, 1 H), 1.75 (br s, 1 H), 1.82-1.86 (m, 1 H), 3.59-3.67 (m, 2 H), 7.07-7.10 (m, 2 H), 7.15-7.20 (m, 1 H), 7.25-7.30 (m, 2 H)
;
13C NMR (100 MHz, CDCl
3) δ: 13.8, 21.2, 25.2, 66.3, 125.6, 125.8, 128.3, 142.5
;
[α]20
D
+82° (EtOH,
c 1.74) [lit.
4
(2R,3R)-cyclopropylmethanol >99% ee
[α]20
D
−92° (EtOH,
c 1.23)]. Anal. Calcd for C
10H
12O: C, 81.04 H, 8.16. Found: C, 81.15; H, 8.30. The enantiomeric excess of the product is determined precisely by GC analylsis of the corresponding
trifluoroacetate ester derivative: To a solution of 10 mg of the crude alcohol in
0.75 mL of pyridine
is added
0.25 mL of trifluoroacetic anhydride (TFAA). After 30 min at room temperature, an additional
0.25 mL of TFAA is added. After 30 min, the reaction mixture is diluted with
5 mL of ether
. This solution was injected directly into the GC (0.5 μL) with the following conditions: Cyclodex G-TA, 0.32 × 30 m; pressure 25 psi; isotherm: 110°C, T
r (minor) 11.5 min, T
r (major) 12.0 min; enantiomeric ratio: 29:1 (93% ee).
35.
If the resulting organic layers are not clear, the combined organic layers should be washed with an additional
50 mL of aqueous saturated ammonium chloride
.
36.
An additional 10-15% of the diol can be recovered from the first aqueous saturated
ammonium chloride extract
(Note 38): the layer is concentrated on a
rotatory evaporator and the white solid is triturated with cold
methanol (30 mL), the mixture is filtered on a Büchner funnel, and the solid is washed with cold
methanol (20 mL). The filtrate is concentrated to ca. 25 mL and treated with
2.5 g of sodium sulfide
(Note 39). The resulting mixture is stirred for 30 min and then filtered on Celite (6 g, 1 cm × 4 cm). The filtrate is concentrated by rotary evaporation, and the residue is purified by flash chromatography on silica gel (75 g, 3.5 cm × 14.5 cm) by dissolving it in
10 mL of 10%
methanol
in
chloroform and eluting with
10%
methanol
in
chloroform. A recrystallization with
dichloromethane and
ethyl acetate give pure material.
37.
The physical properties are identical to those of
(Note 24).
38.
The diol decomposes after a few hours at room temperature in this layer.
39.
Sodium sulfide was purchased from Anachemia Science
and was used without further purification.
Handling and Disposal of Hazardous Chemicals
The procedures in this article are intended for use only by persons with prior training in experimental organic chemistry. All hazardous materials should be handled using the standard procedures for work with chemicals described in references such as "Prudent Practices in the Laboratory" (The National Academies Press, Washington, D.C., 2011 www.nap.edu). All chemical waste should be disposed of in accordance with local regulations. For general guidelines for the management of chemical waste, see Chapter 8 of Prudent Practices.
These procedures must be conducted at one's own risk. Organic Syntheses, Inc., its Editors, and its Board of Directors do not warrant or guarantee the safety of individuals using these procedures and hereby disclaim any liability for any injuries or damages claimed to have resulted from or related in any way to the procedures herein.
3. Discussion
Parts A-D. The preparation of boronic acids by the addition of a Grignard reagent to a trialkyl borate is one of the most convenient and well-established methods involving relatively inexpensive starting materials.
5
6 The carefully monitored addition of
butylmagnesium bromide to
trimethyl borate avoids any complications resulting from overaddition, and relatively good yields of the
butylboronic acid are obtained after acid hydrolysis. Usually, alkylboronic acids are relatively difficult to characterize and to obtain analytically pure,
7
8 because they readily tend to form boroxines (anhydrides) under dehydrating conditions (when heated or when left under reduced pressure). The white solid (
butylboronic acid) is transformed into a colorless oil (tributylboroxine) if dehydration is pushed to completion. Conversely,
butylboronic acid and its boroxine are also readily oxidized by air to generate
1-butanol and
boric acid.
9
10 For these reasons, it is almost impossible to isolate
butylboronic acid without any traces of water, its boroxine or oxidation by-products.
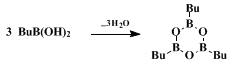
In order to avoid these complications,
butylboronic acid is quickly converted to its air-stable and more robust diethanolamine derivative
1.
11 Complex
1 could be contaminated with some unseparable
diethanolamine if a small excess of
diethanolamine is used in its preparation. However, this has no effect on the efficiency of the synthesis of the chiral
dioxaborolane ligand
3.
Diethanolamine complex
1 reacts quantitatively with a slight excess of
tetramethyltartramide
2 under biphasic conditions to produce the desired chiral
dioxaborolane ligand
3. Ligand
3 is relatively stable, and is neither excessively hygroscopic nor oxygen-sensitive. It must be stored under
argon for longer periods of time. However, the submitters have shown that the enantioselectivities are directly related to the ligand purity. Consequently, it is generally preferable to use freshly prepared ligand for obtaining optimal results.
Part E. The enantioselective cyclopropanation of allylic alcohols using the chiral
dioxaborolane ligand
3 and Zn(CH
2I)
2·DME is a powerful tool for synthesizing three-membered rings. This method is much simpler and produces superior enantiomeric excesses compared to those using other stoichiometric chiral ligands.
12
13
14 The scope of the reaction is wide and a variety of allylic alcohols have been converted into their cyclopropane derivatives in excellent enantiomeric excesses (88-94%).
15
16 It was also shown that polyenes can be cyclopropanated at the allylic alcohol position with excellent chemo- and enantioselectivities.
17 Recently, this reaction has been used in the synthesis of cyclopropane containing natural products.
18
19
20
21
Caution! The previously reported preparation of Zn(CH2I)2 without a complexing additive22 is highly exothermic, and a violent decomposition sometimes occurred. For safety reasons, the use of the Zn(CH2I)2·DME as reported here is mandatory if this reaction is carried out on a =8 mmole scale.23 If the internal temperature during the formation of the reagent is carefully monitored, the procedure reported here is extremely safe even on larger scales.
Note that the structure of Zn(CH2I)2·DME is derived from the stoichiometry of the reactants (Et2Zn, CH2I2, DME). Substantial quantities of IZnCH2I·DME are necessarily formed at the reaction temperature and as a by-product of the cyclopropanation. Another improvement was made in this procedure: the number of equivalents of the reagent has been decreased to 2.0 equiv (vs 5 equiv in the original paper). However, under these conditions that minimize the amount of Et2Zn used but require longer reaction times, the yield of the diol recovery dropped to ca. 50%.
The cyclopropanation of cinnamyl alcohol is a good example of the use of dioxaborolane ligand 3 as chiral additive to synthesize chiral cyclopropanes.
Appendix
Chemical Abstracts Nomenclature (Collective Index Number);
(Registry Number)
(2S,3S)-(+)-(3-Phenycyclopropyl)methanol:
Cyclopropanemethanol, 2-phenyl-,(1S-trans)- (12); (110659-58-0)
Butylmagnesium bromide:
Magnesium, bromobutyl- (8,9); (693-03-8)
Magnesium (8,9); (7439-95-4)
1-Bromobutane:
Butane, 1-bromo- (8,9); (109-65-9)
Isopropyl alcohol:
2-Propanol (8,9); (67-63-0)
1,10-Phenanthroline (8,9); (66-71-7)
Butylboronic acid:
1-Butaneboronic acid (8);
Boronic acid, butyl- (9); (4426-47-5)
Trimethyl borate:
Boric acid, trimethyl ester (8,9); (121-43-7)
Diethanolamine:
Ethanol, 2,2'-iminodi- (8);
Ethanol, 2,2'-iminobis- (9): (111-42-2)
(4R-trans)-2-Butyl-N,N,N',N'-tetramethyl[1,3,2]dioxaborolane-4,5-dicarboxamide:
1,3,2-Dioxaborolane-4,5-dicarboxamide, 2-butyl-N,N,N'N'-tetramethyl-, (4R-trans)- (13); (161344-85-0)
(R,R)-(+)-N,N,N',N'-Tetramethyltartaric acid diamide:
Tartramide, N,N,N'N'-tetramethyl-, (+)- (8);
Butanediamide, 2,3-dihydroxy-N,N,N'N'-tetramethyl-, [R-(R,R)]- (9); (26549-65-5)
Dimethoxyethane:
Ethane, 1,2-dimethoxy- (8,9); (110-71-4)
Diethylzinc:
Zinc, diethyl- (8,9); (557-20-0)
Diiodomethane:
Methane,diiodo- (8,9); (75-11-6)
Cinnamyl alcohol (8);
2-Propen-1-ol, 3-phenyl- (9); (104-54-1)
Hydrogen peroxide (8,9); (7722-84-1)
Sodium sulfite:
Sulfurous acid, disodium salt (8,9); (7757-83-7)
Copper (8,9); (7440-50-8)
Trifluoroacetic anhydride:
Acetic acid, trifluoro-, anhydride (8,9); (407-25-0)
Sodium sulfide (8,9); (1313-82-2)
Copyright © 1921-, Organic Syntheses, Inc. All Rights Reserved