Org. Synth. 1999, 76, 123
DOI: 10.15227/orgsyn.076.0123
METHYL (S)-2-PHTHALIMIDO-4-OXOBUTANOATE
[
2H-Isoindole-2-acetic acid, 1,3-dihydro-1,3-dioxo-α-(2-oxoethyl)-, methyl ester, (S)-
]
Submitted by Patrick Meffre, Philippe Durand, and François Le Goffic
1
.
Checked by David A. Ellis, Yvan Mermet-Bouvier, and David J. Hart.
1. Procedure
A.
Methyl (S)-2-phthalimido-4-methylthiobutanoate
. Into a 2-L, round-bottomed flask fitted with a Dean-Stark apparatus, reflux condenser, and drying tube containing calcium chloride are placed
L-methionine methyl ester hydrochloride (50.0 g, 0.25 mol, (Note 1) and (Note 2)),
phthalic anhydride (37.1 g, 0.25 mol),
triethylamine (100 mL, 0.72 mol), and
toluene (1 L). The mixture is magnetically stirred and heated under reflux for 4.5 hr at which point approximately 4.5 mL of water has separated. The reaction mixture is allowed to cool to room temperature and the precipitated
triethylamine hydrochloride (34 g) is collected by suction filtration. The filtrate is washed with four
300-mL portions of 1 N aqueous hydrochloric acid
followed by three 300-mL portions of water. The organic layer is dried over
magnesium sulfate
, filtered, and the filtrate is concentrated under reduced pressure using a rotary evaporator. The residual oil is placed under reduced pressure for 12 hr at 0.1-0.5 mm, followed by trituration with
200 mL of pentane
to give 59 g (80%) of product as a white solid after collection and drying at room temperature under reduced pressure (mp 37-40°C) (Note 3).
B.
Methyl (S)-2-phthalimido-4-oxobutanoate
. Caution! This reaction must be performed under a well-ventilated hood. Into a 500-mL, two-necked, round-bottomed flask fitted with a gas inlet and a drying tube containing calcium chloride are placed
methyl (S)-2-phthalimido-4-methylthiobutanoate (15.45 g, 52.7 mmol) and
carbon tetrachloride (CCl4) (160 mL)
(Note 4). The mixture is magnetically stirred under nitrogen at room temperature,
N-chlorosuccinimide (7.04 g, 52.7 mmol) is added in one portion, and the reaction mixture is stirred for 2 hr at room temperature. The resulting mixture is filtered through a sintered glass funnel with suction into a 1-L, three-necked, round-bottomed flask, and the collected succinimide (5.2 g) is rinsed with
100 mL of carbon tetrachloride
. The three-necked flask containing the filtrate and rinse is equipped with a gas inlet (Note 5), a stopper, and a cold water condenser. The condenser is equipped with a gas outlet that in turn is connected to a 500-mL Erlenmeyer flask containing 200 mL of sodium hypochlorite solution (commercial bleach) to scavenge volatile sulfur-containing by-products. Water (320 mL) is added in one portion, and nitrogen is bubbled through the solution at a rate of 2.4-3.0 mL/sec for a period of 20 hr at room temperature. The resulting phases are separated using a separatory funnel, and the acidic aqueous phase is extracted with
dichloromethane (2 × 50 mL). The combined organic phases are washed with saturated aqueous sodium bicarbonate (100 mL), and water (100 mL) and then dried over
sodium sulfate
. The solution is filtered, and the filtrate is concentrated at reduced pressure using a rotary evaporator to give 14.0 g of crude product as a pale yellow oil (Note 6). Chromatography of this oil over silica gel
(Note 7) gives a white solid that is triturated at −15°C (ice-salt bath) using
20 mL of diethyl ether
to provide 7.50-9.19 g (54-67%) of analytically pure aldehyde as a white solid (mp 52-103°C) (Note 8), (Note 9) and (Note 10).
2. Notes
1.
Unless stated otherwise, all solvents and reagents were used as purchased (reagent grade) without further purification.
2.
Commercial
L-methionine methyl ester hydrochloride (from Aldrich Chemical Company, Inc.) is used or can be easily prepared using a literature procedure.
2
3.
The properties of
methyl (S)-2-phthalimido-4-methylthiobutanoate follow: mp
37-40°C, lit.
3 mp
33-34°C;
[α]20
D
−41.6° (CHCl
3,
c 1.49), lit.
3
[α]20
D
−46.2° (CHCl
3,
c 1);
1H NMR (200 MHz, CDCl
3) δ 2.02 (s, 3 H), 2.45-2.60 (m, 4 H), 3.69 (s, 3 H), 5.05 (m, 1 H), 7.73 (m, 2 H), 7.81 (m, 2 H)
;
13C NMR (50.3 MHz, CDCl
3) δ 15.2, 28.0, 30.7, 50.7, 52.8, 123.5, 131.7, 134.2, 167.5, 169.5
; MS (CI, DCI-NH
3): 294 (M+H)
+
, 311 (M+NH
4)
+
. Anal. Calcd. for C
14H
15NO
4S: C, 57.33; H, 5.12; N, 4.78. Found: C, 57.29; H, 5.19; N, 4.72. This material was shown to have 96-98% ee based on HPLC analysis over a
25-cm × 0.46-cm Chiracel OJ column using
hexane-isopropyl alcohol (98:2) as eluant and a flow rate of approximately 1 mL min
−1. The S-isomer elutes with a retention time of approximately 47 min, while the R-isomer has a retention time of approximately 66 min.
4.
Carbon tetrachloride is stored over 4 å molecular sieves before use. The chlorination is performed using oven-dried glassware quickly assembled before use.
5.
The gas inlet consists of an
18-gauge needle passed through a rubber septum reaching to the bottom of the 1-L reaction vessel.
6.
TLC analysis using silica gel 60-F254 (0.2-mm thick on
aluminum sheets) and visualized under UV lamp or with
ammonium molybdate-based reagent [36 mL of
H2O, 4 mL of concd H
2SO
4, 1 g of (NH
4)
6Mo
7O
24·4H
2O, and 0.4 g of Ce(SO
4)
2] shows the presence of two spots with R
f = 0.3 and R
f = 0.15 (cyclohexane/ethyl
acetate, 2:1).
7.
The oil is dissolved in 20 mL of cyclohexane-ethyl
acetate (3:1) and 10 mL of
dichloromethane, applied to a 25-cm high × 8-cm wide column of Merck silica gel 60 (230-400 mesh), and eluted using flash chromatography.
4 The column is eluted with 4 L of cyclohexane-ethyl
acetate (3:1) followed by 3 L of cyclohexane-ethyl
acetate (1:1). After a forerun of 2.3 L is collected, early fractions containing the higher R
f material
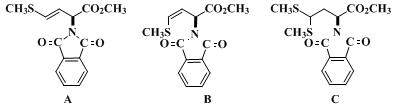
(Note 6) are obtained (2.2 g). This material is a mixture of starting material, alkenes
A and
B, thioacetal
C, and other material. Elution of the aldehyde begins after about 4 L of eluant have been passed through the column.
8.
The properties of
methyl (S)-2-phthalimido-4-oxobutanoate follow:
[α]20
D
−44.4° (CHCl
3,
c 1.49);
1H NMR (200 MHz, CDCl
3) δ 3.26 (ddd, 1 H, J = 18.4, 7.7, 0.8), 3.55 (ddd, 1 H, J = 18.4, 6.0, 0.8 ), 3.74 (s, 3 H), 5.51 (dd, 1 H, J = 7.7, 6.0), 7.77 (m, 2 H), 7.90 (m, 2 H), 9.75 (t, 1 H, J = 0.8)
;
13C NMR (50.3 MHz, CDCl
3) δ 42.8, 45.9, 53.1, 123.6, 131.5, 134.3, 167.1, 168.8, 197.4 (CHO)
; MS (CI, DCI-NH
3): 262 (M+H)
+
, 279 (M+NH
4)
+
; IR (neat) cm
−1: 1720, 1750, 1780, 2720, 2820
. Anal. Calcd. for C
13H
11NO
5: C, 59.77; H, 4.24; N, 5.36. Found: C, 59.47; H, 4.49; N, 5.32.
9.
The (R)-aldehyde could be prepared from the
hydrochloride salt of D-methionine methyl ester using a similar two-step procedure.
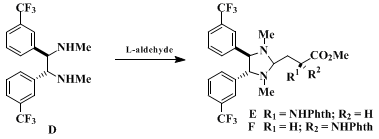
10.
This material was analytically pure, but it melted over a broad range because of the presence of a mixture of enantiomers. This material was shown to have approximately 90-92% ee using the following procedure:
5 To a stirred solution of
(1R,2R)-(+)-N,N'-dimethyl-1,2-bis(3-trifluoromethyl)phenyl-1,2-ethanediamine (D) (50 mg, 0.13 mmol) in
10 mL of ether
containing 1 g of 4 Å molecular sieves at room temperature under an
argon atmosphere is added a solution of the (
L)-aldehyde (35 mg, 0.13 mmol) in
10 mL of ether
. The mixture is stirred at room temperture for 2 hr, whereupon TLC analysis (
silica gel,
cyclohexane-
ethyl acetate, 2:1) indicates that the reaction is complete. The solution is filtered, and the filtrate is concentrated to give a mixture of diastereomeric imidazolidines (
E and
F) whose ratio is best determined by integration of the methine signals (
1H NMR in
benzene-d6
at 500 MHz) that appear at α 3.68 in diastereomer
E and at δ 3.94 in diastereomer
F.
Care should be taken to take NMR spectra with a pulse delay long enough to ensure that relaxation of all nuclei occurs. The checkers found that integration of other signals in the 1H and 19F spectra that were less well resolved gave lower % ee values. They also found that integration of the aforementioned signals on lower field instruments (200 and 300 MHz) gave lower % ee values because of partial overlap of signals.
Handling and Disposal of Hazardous Chemicals
The procedures in this article are intended for use only by persons with prior training in experimental organic chemistry. All hazardous materials should be handled using the standard procedures for work with chemicals described in references such as "Prudent Practices in the Laboratory" (The National Academies Press, Washington, D.C., 2011 www.nap.edu). All chemical waste should be disposed of in accordance with local regulations. For general guidelines for the management of chemical waste, see Chapter 8 of Prudent Practices.
These procedures must be conducted at one's own risk. Organic Syntheses, Inc., its Editors, and its Board of Directors do not warrant or guarantee the safety of individuals using these procedures and hereby disclaim any liability for any injuries or damages claimed to have resulted from or related in any way to the procedures herein.
3. Discussion
The synthetic utility of amino acids for the generation of chiral reagents, intermediates, and final products including amino acids is well documented.
6
7
8 Derivatives of
(S)-2-amino-4-oxobutyric acid (
aspartic acid β-semialdehyde,
3-formylalanine) are useful chiral intermediates for the synthesis of biologically relevant molecules of wide interest including:
nicotinamine and analogues, iron chelating agents,
9
10
11
12
13 naturally occurring unusual α-amino acids,
14
15
16
17
18
19
20
21 serine-phosphate peptide isosteres,
22
23
24 penicillin or cephalosporin analogues,
25
26 and compounds used in some enzyme studies.
27
28
29 These compounds have been synthesized from the expensive
allylglycine via ozonolysis or oxidative cleavage,
9,10,27,28 from
aspartic acid via reduction of the acid side chain to give homoserine derivatives,
11,14,15,22,23,24 from
methionine via a homoserine derivative
16 and subsequent oxidation to the aldehyde, or directly from the expensive
homoserine.
12,13,17,18,25
26,29 Direct reduction of reactive aspartic acid derivatives also led to the aldehyde.
19,20,21 These strategies often suffer from low yields because of the number of steps or from the use of expensive or impractical starting materials or reagents. The synthesis of
ethyl (S)-2-phthalimido-4-oxobutanoate has been described
30 from
L-methionine using corrosive
sulfuryl chloride as the chlorinating agent followed by a time-consuming hydrolysis, but partially racemized compounds are obtained, probably because of the harsh experimental conditions. The procedure described here provides material of greater than 90% ee using inexpensive and easily handled reagents.
In the procedure presented here, the phthaloyl group is chosen as an amino protecting group to avoid internal amine participation.
31 Amine diprotection is necessary since the same reaction conducted on the N-benzyloxycarbonyl (NHCbz) and the N-tert-butyloxycarbonyl (NHBoc) analogues did not led to clean α-chlorination but rather to unidentified products. This protection was performed using a standard procedure.
32
The second step is a Pummerer-like reaction using easily handled
N-chlorosuccinimide (NCS)
33,34
35 in
carbon tetrachloride. α-Chlorination was chosen because of its regioselectivity towards an α-methylene compared with an α-methyl group.
36
37 NMR analysis of the crude mixture of chlorinated intermediates (mixture of isomers)
30,33 indicates that chlorination occurs mainly at the methylene group, although some vinyl sulfide products are also present at this stage of the reaction.
Succinimide formed in the reaction is not soluble in CCl
4, and floats on the surface (unlike
N-chlorosuccinimide) indicating completion of the reaction; it is easily separated by filtration. Hydrolysis of the transient chlorinated intermediates is then immediately performed using water and bubbling
nitrogen to help remove
methanethiol from the reaction mixture. Hydrolysis using HgCl
2/CdCO
3/
H2O,
38 NCS/AgNO
3/
acetonitrile,
39 or CuCl
2·2H
2O/CuO/H
2O/acetone
40 leads to lower yields.
Appendix
Chemical Abstracts Nomenclature (Collective Index Number);
(Registry Number)
Methyl (S)-2-phthalimido-4-oxobutanoate:
2H-Isoindole-2-acetic acid, 1,3-dihydro-1,3-dioxo-α-(2-oxoethyl)-, methyl ester, (S)- (12); (137278-36-5)
Methyl (S)-2-phthalimido-4-methylthiobutanoate:
2H-Isoindole-2-acetic acid, 1,3-dihydro-α-[2-(methylthio)ethyl]-1,3-dioxo-, methyl ester, (S)- (9); (39739-05-4)
L-Methionine methyl ester hydrochloride:
Methionine, methyl ester, hydrochloride, L- (8);
L-Methionine, methyl ester, hydrochloride (9); (2491-18-1)
Phthalic anhydride (8);
1,3-Isobenzofurandione (9); (85-44-9)
Triethylamine (8);
Ethanamine, N,N-diethyl- (9); (121-44-8)
Toluene (8);
Benzene, methyl- (9); (108-88-3)
Carbon tetrachloride: CANCER SUSPECT AGENT (8);
Methane, tetrachloro- (9); (56-23-5)
N-Chlorosuccinimide:
Succinimide, N-chloro- (8);
2,5-Pyrrolidinedione, 1-chloro- (9); (128-09-6)
Sodium hypochlorite solution:
Hypochlorous acid, sodium salt (8,9); (7681-52-9)
Copyright © 1921-, Organic Syntheses, Inc. All Rights Reserved