Org. Synth. 1925, 5, 59
DOI: 10.15227/orgsyn.005.0059
ETHYL OXALATE
[Oxalic acid, ethyl ester]
Submitted by H. T. Clarke and Anne W. Davis.
Checked by Roger Adams and W. B. Burnett.
1. Procedure
In a 5-l. flask are placed 1 kg. (7.9 moles) of crystallized (hydrated) oxalic acid, 1.66 kg. (2034 cc.) of 95 per cent ethyl alcohol, and 1.33 kg. (887 cc.) of carbon tetrachloride. The flask is then fitted with a fractionating column, 1 meter long, to which are attached a condenser and an automatic separator so arranged that the lighter liquid flows off to a receiver (Fig. 13) (Note 1). The heavier liquid flows through a tower of anhydrous potassium carbonate, and then returns to the reaction flask. The bottom of the tower is connected with a small separatory funnel through which any potassium carbonate solution, which flows from the solid in the tower, may be withdrawn from time to time.
The mixture in the flask is slowly distilled (Note 2). As soon as about 500 cc. of the lighter liquid has collected, it is placed in a fractionating apparatus and distilled, the material which boils up to 79° being collected separately. This fraction, which consists principally of alcohol, with a little carbon tetrachloride and moisture, is dried with potassium carbonate and returned to the reaction mixture (Note 3). The higher fractions are redistilled.
The above process is continued until the distillate no longer separates into two phases (about twenty-seven hours). The liquid in the flask is then distilled with the use of a column until the temperature of the vapor reaches 85°; the residue is then distilled under reduced pressure, and the fraction which boils at 106–107°/25 mm. is collected. The yield is 920–960 g. of a colorless liquid (80–83 per cent of the theoretical amount) (Note 4).
2. Notes
1.
The apparatus shown in Fig. 13 may be somewhat more simply constructed by using rubber connections in several places, thus eliminating
Fig. 13.
a certain amount of glass blowing, and making a more flexible piece of apparatus. The side arm of the separator may be made with two rubber connections—one above and one below the tube leading to the potassium carbonate tube. The long return tube to the flask may be constructed with a rubber joint very near the carbonate tube and one near the flask.
2.
Water,
ethyl alcohol, and
carbon tetrachloride form a ternary mixture boiling at about 61°. This vapor mixture, on condensation, separates into two phases; the heavier liquid consists of
carbon tetrachloride and alcohol with only small amounts of water; the lighter liquid consists of approximately
65 per cent alcohol, 25 per cent water, and
10 per cent carbon tetrachloride. By taking advantage of this fact, it is possible to conduct the esterification at a temperature so low that the
ethyl hydrogen oxalate first formed does not decompose into
ethyl formate and other products, as it does when the customary methods of esterification are employed.
The reaction may be carried out somewhat more expeditiously if the
oxalic acid is dehydrated independently
(p. 421) before it is mixed with the alcohol; indeed, it is also possible to remove the bulk of the water from the alcohol itself by a similar method, before mixing it with the
oxalic acid. However, since water is formed during the esterification, little is gained by this procedure.
3.
It is not absolutely necessary to remove the last traces of water from the
alcohol-carbon tetrachloride layer by means of
potassium carbonate before returning it to the reaction mixture; this process is, however, so simple and requires so little attention that there is no doubt that it is of material aid in cutting down the time of operation. The advantages of using crystallized
oxalic acid and commercial
95 per cent alcohol, instead of the anhydrous reagents, are obvious. When technical
oxalic acid is used, the yields are usually smaller by
5–10 per cent.
4.
Ethyl oxalate can also be prepared in about the same yield by the esterification procedure described in Org. Syn.
10, 48.
Submitted by Joseph Kenyon
Checked by C. S. Marvel and A. B. Adams.
1. Procedure
In a
large evaporating dish is placed
252 g. (2 moles) of crystalline oxalic acid. The acid is heated on a
steam bath for six to eight hours, with occasional stirring, until all the water of crystallization (72 g.) has been expelled
(Note 1). The
oxalic acid, which is almost anhydrous (weighing approximately 180 g.), is placed in a
1.5-l. round-bottomed flask, A, containing
500 cc. of absolute alcohol (p. 249) and fitted up as shown in Fig. 14.
Fig. 14.
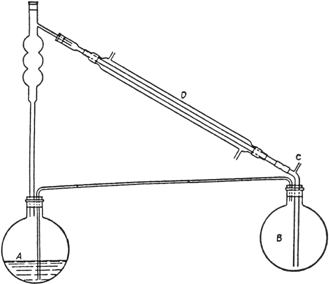
The flask A is heated by means of an oil bath maintained at 120–125° (Note 2); the mixed vapors of alcohol and water, passing through the fractionating column, are condensed in D, and the moist alcohol is delivered under the surface of the alcohol contained in flask B. In flask B are placed 250 cc. of absolute alcohol and about 200 g. of freshly ignited potassium carbonate. Flask B is heated in an oil bath maintained at about 95–100°. The moist alcohol delivered to the flask B is dried by the potassium carbonate and subsequently returned as vapor under the surface of the liquid in flask A. The tube C, which is provided with a calcium chloride tube and is open to the air, acts as a vent for the otherwise closed system. The reaction is run for about five hours. The excess alcohol is then distilled, the residue of ethyl oxalate is distilled under reduced pressure, and the fraction boiling at 98–101°/21 mm. is collected. The yield is 234–264 g. (80–90 per cent of the theoretical amount) (Note 3).
2. Notes
1.
The
oxalic acid may be dehydrated by the procedures described on
p. 421.
2.
The temperature varies slightly with the length of the fractionating column. This temperature was noted when an ordinary
two-bulb column (about 30 cm. long) was used.
3.
A similar procedure may be used for the preparation of
methyl oxalate. Instead of distilling this
ester, it is better to cool the solution in an
ice bath and separate the crystals of
methyl oxalate from the mother liquors in a
basket centrifuge. The product thus obtained from
252 g. of crystallized oxalic acid weighs
120–125 g. Upon concentrating the mother liquors, cooling, and again centrifuging the mixture, a further crop of crystals weighing
30–35 g. may be obtained. Thus the total yield of
methyl oxalate is
150–160 g. (
63–67 per cent of the theoretical amount).
Another method for the preparation of methyl oxalate is described in Org. Syn. 10, 70.
3. Discussion
Ethyl oxalate can be prepared by distilling a mixture of anhydrous
oxalic acid and absolute
alcohol, the vapor of absolute
alcohol being passed simultaneously into the mixture;
1 and by adding sufficient alcohol to remove as the azeotropic mixture all the water formed in the reaction and then heating the mixture until the water is removed.
2 A modification of this method is the use of a dehydrating agent in the system,
3 as illustrated in (
B) of the procedure. Good yields have been obtained by saturating a mixture of crystallized
oxalic acid and alcohol with
hydrogen chloride, removal of the alcohol and water by distillation under reduced pressure, and repetition of the treatment with alcohol and
hydrogen chloride;
4 and by stirring for several hours at room temperature a mixture of anhydrous
oxalic acid,
95 per cent ethyl alcohol,
sulfuric acid, and
benzene.
5
Other procedures which have been employed include: The use of Twitchell's reagent as a catalyst;
6 the distillation of a mixture of anhydrous
oxalic acid, absolute
alcohol,
sulfuric acid and
toluene;
7 and the distillation of a mixture of
oxalic acid, absolute
alcohol, and a liquid which forms a ternary mixture with the alcohol and water, distillation being arranged so that the vapors are condensed and returned to the reaction mixture after passing through a
thimble containing calcium carbide.
8This preparation is referenced from:
Appendix
Chemical Abstracts Nomenclature (Collective Index Number);
(Registry Number)
ester
ethyl hydrogen oxalate
alcohol-carbon tetrachloride
ethyl alcohol,
alcohol (64-17-5)
potassium carbonate (584-08-7)
sulfuric acid (7664-93-9)
hydrogen chloride (7647-01-0)
Benzene (71-43-2)
carbon tetrachloride (56-23-5)
Oxalic acid (144-62-7)
toluene (108-88-3)
Ethyl oxalate,
Oxalic acid, ethyl ester
ethyl formate (109-94-4)
Methyl oxalate
Copyright © 1921-, Organic Syntheses, Inc. All Rights Reserved