Org. Synth. 1921, 1, 61
DOI: 10.15227/orgsyn.001.0061
METHYL-n-HEXYLCARBINOL
[2-Octanol]
Submitted by Roger Adams and C. S. Marvel.
Checked by Oliver Kamm and W. S. Marloth.
1. Procedure
For the most satisfactory preparation of methylhexylcarbinol from castor oil, in moderately large-sized runs, a copper vessel should be used (B). Since such a vessel is often not available in the average laboratory, directions are also included using an ordinary can (A), although in such cases the yields are much lower, and relatively smaller quantities of materials are used.
(A) Preparation in Tin Vessel.—In a large dish pan, 1900 g. (6.4 moles) of No. AAA castor oil (Note 1) is treated with occasional stirring with a solution of 500 g. (12.5 moles) of sodium hydroxide in 300 cc. of water. A reaction takes place, some heat is developed, and within ten or fifteen minutes a very hard solid soap is produced. This soap is broken up into small pieces and placed in a 3-gallon (12-l.) can (Note 2). In this preparation, a petroleum ether can was used, but any kind available would be just as satisfactory. The container is now fitted with an efficient reflux condenser, and it is then heated over a ring burner as long as hydrogen is evolved. The heating is regulated to produce a fairly rapid evolution of gas as shown by leading a tube from the top of the condenser into a small beaker of water. The time required for the complete evolution of the hydrogen is about nine to ten hours.
If the above reaction is not completed at one time and intermediate cooling is allowed, the reaction mixture sets to a solid mass and certain precautions must be taken before proceeding further. Either before heating is renewed the mass must be punched full of holes by means of an iron rod, or when the heating is started care must be taken to heat the upper part of the can first and gradually to approach the bottom. If the under part of the solid soap were heated directly, it would decompose before the upper part, and the hydrogen, being unable to escape, would cause the can to burst.
The condenser is now set downward for distillation and the heating is continued. If intermediate cooling is allowed, one of the precautions mentioned above must be observed before starting the distillation. The alcohol distils over with the water and is separated from time to time, the water being returned to the distillation can by means of a separatory funnel inserted through a rubber stopper in the top of the can. The addition of this water must be very slow in order to prevent foaming (Note 3). The heating should be regulated so that the distillate is coming over in rapid drops, not in a stream. If desired, the same sort of gravity separator described below under the large-sized experiment (B) may be used in this small run. About twelve hours are required for this distillation (Note 4). Toward the end, high-boiling products are obtained and considerable gas is liberated. Difficulty is almost always encountered during this part of the preparation because the long heating causes the solder in the bottom edge of the can to melt and leaks thus develop which allow a certain amount of soap to run out. The distillation need not be stopped, because the soap fills the holes, but under these conditions the temperature cannot be maintained at a point high enough for the complete distillation without material loss from increased leakage. The upper layer of crude alcohol is separated from the water and fractionated. The portion boiling from 100–120° is chiefly water with a little alcohol in it. The second fraction between 120° and 175° contains a considerable quantity of ketones along with the alcohol (Note 4). The main portion distilling between 175° and 185° is the methylhexylcarbinol. Upon refractionation,
190–200 g. (23–25 per cent of the theoretical amount) of alcohol boiling at 175–180° is obtained.
This product is satisfactory for most purposes. If, however, a very pure product is desired, this may be made by shaking carefully the alcohol with 15 per cent sodium bisulfite solution, separating the alcohol, and steam distilling it from alkaline solution. The alcohol is finally distilled, and the fraction boiling at 177–179° is collected. If saturated sodium bisulfite is employed, a crystalline material which contains alcohol is formed and causes difficulty in the separation (Note 5).
Two runs of 6 kg. of castor oil with the same relative proportions of alkali and water were made in 8-gallon (30-l.) cans. The yields of pure alcohol were 650 g. and 730 g. (25.8 per cent and 29 per cent, respectively, of the theoretical amount on the basis of pure ester).
(
B)
Preparation in Copper Vessel.—To the cover of a
12-gallon (45-l.) copper kettle (Note 6) is fitted an apparatus to which a
continuous separator may be attached (see Fig. 20).
Fig. 20.
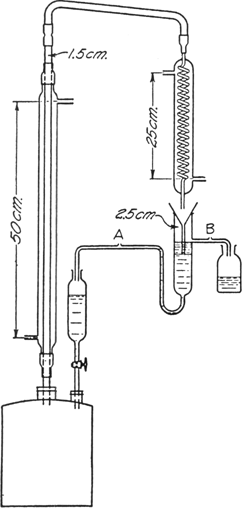 |
This apparatus consists of an
upright water condenser through the jacket of which steam is passed; a
connecting tube leads from the top of this condenser to a second
spiral condenser the bottom of which extends into a gravity separator. This allows the alcohol to run off into a
receiver and the heavier aqueous layer to return through a separatory funnel to the copper kettle. A few essential points in the apparatus should be indicated: it is necessary that the return arm of the gravity separator be below the tube through which the alcohol flows into the receiver; the lower end of the funnel must be below the return side arm in order to prevent water being carried along with the
methylhexylcarbinol; the capillaries A and B are necessary to prevent siphoning in the respective tubes.
A soap is made as already described (
A) from 11.2 kg. (37.6 moles) of No. AAA castor oil
(Note 1) and
2900 g. (73 moles of sodium hydroxide in 5800 g. of water
(Note 7). This is broken up, placed in the kettle, and gradually heated. Much gas is evolved, and care should be taken not to lose alcohol, particularly at the beginning. The separator is not adjusted and the
stopcock of the separatory funnel is kept closed until about 3 l. of distillate is collected; any small amount of alcohol is separated and the water discarded
(Note 7). The separator is now put in place to return the water to the kettle. The separatory funnel is allowed to fill with liquid and then the stopcock regulated so that the liquid flows back to the copper vessel as fast as it distils over. A
return tube similar to the one illustrated in
Fig. 22, p. 422, may be used, but in making
methylhexylcarbinol the separatory funnel has the advantage that, if foaming occurs in the
reaction flask, further addition of water can be immediately and easily stopped
(Note 3).
The distillation is continued until no more alcohol comes over, a process which requires forty-eight to seventy-two hours of continuous heating. Superheating must be avoided, since foaming will then occur and may cause considerable difficulty. It is therefore advisable to adjust the heating so that the distillate comes over in rapid drops but not in a stream. If cooling is allowed to take place before the distillation is complete, it is advisable to break up the reaction cake before heating again, or to start the heating at the top of the cake and to approach the bottom gradually (Note 8). The crude product obtained is very much lighter in color than that produced when cans are used (A), and when it is distilled only very small fractions of low- or high-boiling material result. Moreover, the amount of ketones is small. The yields of the redistilled fraction boiling at 175–180° in three successive experiments were 1854 g., 1955 g., and 1894 g. (40–42 per cent of the theoretical amount), quantities about twice those obtained when cans are used (A). The time that one actually devotes to the production of methylhexylcarbinol is not great, but the total time necessary for carrying out a large experiment is almost a week.
2. Notes
1.
The best grade of castor oil gives much better results than the poorer grades.
2.
The
container which is used in the production of
methylhexylcarbinol should not be filled more than one-half with the reaction mixture, since foaming occurs to a considerable extent during the heating.
3.
Upon adding to the reaction mixture the water which distils over with the alcohol, the special precaution must be taken of making the addition slowly or else the contact of the cold liquid with the hot reaction mass causes very bad foaming. If the distillation is too rapid, superheating occurs, and excessive foaming is liable to take place.
4.
It is probable that stirring during the decomposition of the soap would increase the speed of the reaction and decrease the by-products.
5.
It is suggested that the
methylhexylcarbinol be purified as follows: The
190–200 g. of material boiling at
175–180° and
20 g. of phenylhydrazine are warmed on the
steam bath for one hour and then steam distilled; the upper oily layer in the distillate is first washed with dilute
hydrochloric acid, and then stirred and refluxed with
20 g. of stannous chloride and
35 cc. of hydrochloric acid for about one hour. During this time the material becomes very dark in color. The cooled oily layer is washed once with water, steam distilled, the distillate washed with water and
dilute
potassium carbonate, and finally distilled under reduced pressure after drying over
potassium carbonate (W. W. Hartman, private communication).
6.
It is worthy of special note that the yield is much larger when the reaction is carried out in a copper vessel that is capable of being heated to a high temperature without leaking. An iron vessel cannot be substituted for
copper. Several runs in
iron gave about the same weight of crude product, but this always contained high and low fractions that did not appear when
copper was used.
7.
It will be noticed that 5800 g. of water is used in the large runs (
B) and then 3000 g. is distilled off and discarded before the alcohol begins to come over rapidly. This quantity of water helps in the formation of the soap, but it is not absolutely necessary; only 2000 or 2500 g. may be used, under which conditions the alcohol begins to come over immediately with the water.
8.
After all the
methylhexylcarbinol has been distilled from the reaction mixture, the residue in the copper vessel should not be allowed to cool directly, as a solid cake that will have to be chiseled out will be formed. After the heating is stopped, it is advisable to add water to the hot residue, very slowly at first, then, as the kettle cools, more rapidly. While still fairly hot, however, the mixture is poured into a
crock and allowed to cool.
3. Discussion
Methylhexylcarbinol can be prepared by the action of
methylmagnesium iodide on
heptaldehyde,
1 but it is less expensively obtained in a state of purity satisfactory for most purposes by heating castor oil with
sodium hydroxide.
12 This gives
sodium ricinoleate which upon fusion with
caustic soda yields
methylhexylcarbinol.
This preparation is referenced from:
Appendix
Chemical Abstracts Nomenclature (Collective Index Number);
(Registry Number)
caustic soda
ketones
potassium carbonate (584-08-7)
hydrochloric acid (7647-01-0)
hydrogen (1333-74-0)
sodium hydroxide (1310-73-2)
iron (7439-89-6)
Phenylhydrazine (100-63-0)
stannous chloride
sodium bisulfite (7631-90-5)
copper (7440-50-8)
methylmagnesium iodide (917-64-6)
2-Octanol,
Methylhexylcarbinol,
METHYL-n-HEXYLCARBINOL (123-96-6)
sodium ricinoleate (5323-95-5)
heptaldehyde (111-71-7)
Copyright © 1921-, Organic Syntheses, Inc. All Rights Reserved