Org. Synth. 1922, 2, 47
DOI: 10.15227/orgsyn.002.0047
METHYL RED
Submitted by H. T. Clarke and W. R. Kirner.
Checked by Roger Adams and J. B. Davis.
1. Procedure
Six hundred and eighty-five grams (4.75 moles) of technical anthranilic acid (generally about 95 per cent pure) (Note 1) is dissolved in 1.5 l. of water and 500 cc. of concentrated hydrochloric acid (sp. gr. 1.17) (Note 2), by heating. The insoluble dark impurity present in small amounts is filtered off, and the filtrate is transferred to a 16-l. crock and chilled with stirring. It is then mixed with a mush of 2.5 kg. of ice and 750 cc. of concentrated hydrochloric acid. The crock is cooled externally with ice, and the contents stirred continuously. When the temperature reaches about 3°, a filtered solution of 360 g. (5 moles) of sodium nitrite in 700 cc. of water is dropped in slowly, through a long capillary tube reaching below the surface of the liquid (Note 3), until a faint but permanent reaction to starch-potassium iodide paper is obtained; the temperature is kept between 3 and 5° (Note 4). This operation requires all but about 30 cc. of the nitrite solution and occupies one and one-half to two hours.
To the solution of the diazonium salt is now added 848 g. (885 cc., 7 moles) of dimethylaniline; this may be done rapidly, as the temperature does not rise appreciably. Stirring is continued for one hour, the temperature being kept at 5°. Five hundred cubic centimeters of a filtered solution of 680 g. (8.3 moles) of crystallized sodium acetate diluted to 1200 cc. is then added, and the stirring continued for four hours. If a foamy solid rises to the surface during this time and refuses to become incorporated by the stirrer, a few drops of ethyl acetate may be added to reduce the foam. The mixture is allowed to stand overnight in an ice bath which is well insulated by several thicknesses of burlap; the temperature must be kept below 7° to get a good yield of product. The remainder of the sodium acetate solution is then added with stirring, and, after the mixture has been stirred for an additional period of one to three hours, the temperature is allowed to rise slowly to 20–25° in the course of twenty-four hours. Just enough sodium hydroxide solution is then added, with stirring, to cause the mixture to have a distinct odor of dimethylaniline (about 240 cc. of a 40 per cent solution is generally required), and the mixture is allowed to stand for forty-eight hours or longer at room temperature (20–25°) (Note 5).
The solid is then filtered off, washed first with water, then with 400 cc. of 10 per cent acetic acid (to remove the dimethylaniline), and finally with distilled water. The last filtrate is generally pale pink. The solid is sucked as dry as possible, spread out on a tray in order to allow most of the water to evaporate (fifteen to twenty hours), and then suspended in 4 l. of methyl alcohol in a 12-l. flask. This mixture is stirred on the steam bath under a reflux condenser for one to two hours, allowed to cool slowly, and then chilled in an ice bath and filtered. The solid product is washed with a second 4 l. of cold methyl alcohol (Note 6). After being dried in air, it varies in weight from 820 to 870 g.
The product is extracted with boiling
toluene in the following manner: 150 g. is placed in a
fluted filter paper of 29-cm. diameter in a
25-cm. glass funnel which passes through the
cork of a
2-l. flat-bottomed conical flask containing
1250 cc. of toluene (Fig. 21).
Fig. 21.
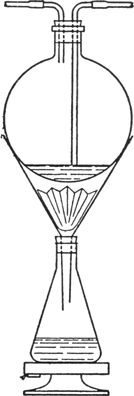 |
The flask is heated on an
electric stove, and a
12-l. round-bottomed flask, clamped to a
ring stand, is placed on the funnel to act as a condenser, cold water being run through the flask. The
toluene is boiled until the condensed liquid runs through almost colorless (this requires from four to ten hours). The heating is then discontinued, and, as soon as the liquid ceases to boil, the flask is removed to a bath containing water at 90–100°; the level of the water should be slightly above the level of the liquid in the flask. This arrangement permits the temperature to fall slowly so that large crystals are obtained. In the meantime a second conical flask containing
1250 cc. of toluene is attached to the funnel, and a new charge of
150 g. of crude methyl red is placed in the paper. The process is repeated until all the crude material has been so treated. When extraction is complete it is found that a certain amount of black amorphous insoluble matter remains on the filter; this is discarded. The crystals of
methyl red (Note 7) are filtered off and washed with a little
toluene. The weight of pure material is
755–805 g. The mother liquors are concentrated to one-fourth of their volume, and the crystals which separate on cooling are recrystallized from fresh
toluene. The recovered
toluene can, of course, be employed again. The total yield of pure
methyl red is
790–840 g. (
62–66 per cent of the theoretical amount)
(Note 8). It melts at
181–182°.
The watery mother liquors from the crude methyl red are rendered alkaline with sodium hydroxide and distilled until no more dimethylaniline passes over. In this way 250 to 400 g. of moist dimethylaniline is recovered.
2. Notes
1.
It is advisable to titrate the crude
anthranilic acid with standard alkali and
phenolphthalein before starting the experiment.
2.
The amount of
hydrochloric acid indicated must not be reduced; otherwise,
diazoamino compounds are formed.
3.
The use of a capillary tube for the addition of
sodium nitrite prevents loss of
nitrous acid by local reaction at the surface of the acid solution. The tube should not be tightly connected to the
dropping funnel, but should be so arranged that air is sucked through with every drop. In this way, the entrance of the acid liquor into the capillary is prevented.
4.
It is essential to keep the temperature low while unreacted
diazobenzoic acid remains in solution, in order to avoid decomposition. If this precaution is not taken, the yields are considerably diminished, through the formation of tarry by-products.
5.
The formation of the azo compound takes place slowly on the addition of the
dimethylaniline, but the speed of the reaction is greatly increased when the
hydrogen-ion concentration is lowered by the addition of the
sodium acetate. It is nevertheless necessary to allow the reaction mixture to stand a long time; if the product is filtered off after only twenty-four hours, a further quantity of dye will separate from the filtrate on standing. The
hydrochloride of methyl red is only sparingly soluble in cold water, and is apt to separate in blue needles if the acidity is not sufficiently reduced.
6.
The alcoholic filtrate, obtained on digesting and washing the crude
methyl red, contains a more soluble red by-product which gives a brownish-yellow solution in alkali. The
methyl alcohol may be recovered with very little loss by distillation; it is, however, impracticable to attempt to recover any
methyl red from the residue, owing to the tarry nature of the by-product. The proportion of this by-product is greatly increased if the temperature of the mixture is allowed to rise too soon after the addition of the
sodium acetate.
7.
Methyl red is described as crystallizing in needles from glacial
acetic acid; on recrystallization from
toluene it separates in plates.
When the methyl red is crystallized from toluene, it sometimes separates in the form of bright-red lumps, probably on account of too rapid crystallization. Under these conditions it is advisable to crystallize again, using a somewhat larger amount of toluene. The sodium salt may be prepared by dissolving the crude product in an equal weight of 35 per cent sodium hydroxide which has been diluted to 3 or 4 liters, filtering, and evaporating under diminished pressure. The isolation of methyl red as the sodium salt avoids the toluene extraction, and yields a water-soluble product (orange leaflets) which is very convenient for use as an indicator.
8.
In checking these directions, an
80 per cent anthranilic acid was used; it gave a correspondingly lower yield of
methyl red (
650–700 g.).
3. Discussion
Methyl red can be prepared by diazotization of
anthranilic acid in alcoholic solution, the product being allowed to react with
dimethylaniline in the same solvent,
1 or both operations can be carried out in aqueous solution.
2
This preparation is referenced from:
- Org. Syn. Coll. Vol. 1, 140
- Org. Syn. Coll. Vol. 1, 149
- Org. Syn. Coll. Vol. 1, 207
- Org. Syn. Coll. Vol. 1, 228
- Org. Syn. Coll. Vol. 1, 335
- Org. Syn. Coll. Vol. 1, 336
- Org. Syn. Coll. Vol. 1, 538
- Org. Syn. Coll. Vol. 2, 580
- Org. Syn. Coll. Vol. 3, 310
- Org. Syn. Coll. Vol. 3, 822
Appendix
Chemical Abstracts Nomenclature (Collective Index Number);
(Registry Number)
Methyl Red
diazoamino compounds
hydrochloride of methyl red
hydrochloric acid (7647-01-0)
acetic acid (64-19-7)
ethyl acetate (141-78-6)
methyl alcohol (67-56-1)
sodium acetate (127-09-3)
sodium hydroxide (1310-73-2)
sodium nitrite (7632-00-0)
nitrous acid (7782-77-6)
nitrite (14797-65-0)
toluene (108-88-3)
dimethylaniline (121-69-7)
Anthranilic Acid (118-92-3)
phenolphthalein (77-09-8)
diazobenzoic acid
hydrogen-ion
Copyright © 1921-, Organic Syntheses, Inc. All Rights Reserved