Org. Synth. 1929, 9, 84
DOI: 10.15227/orgsyn.009.0084
ac-TETRAHYDRO-β-NAPHTHYLAMINE
[2-Naphthylamine, 1,2,3,4-tetrahydro-]
Submitted by E. B. H. Waser and H. Möllering.
Checked by Roger Adams and W. W. Moyer.
1. Procedure
A 1-l. wide-necked, round-bottomed flask is set into an iron dish of about 1.3–1.5 l. capacity and the space between is filled with fine sand so that the flask rests about 1 cm. from the bottom of the dish (Note 1). In the flask is placed 26 g. (1.1 atoms) (twice the theoretical amount) of sodium cut into thin slices (Note 2), and the flask is then closed with a tight-fitting cork stopper through which is inserted a separatory funnel (Note 3) and a long, wide, Liebig condenser through the jacket of which is passed downward a rapid current of air (Note 4).
While this set-up is being prepared a solution of 20 g. (0.14 mole) of β-naphthylamine in 250 cc. of isoamyl alcohol (dried by distilling off a moist forerun) is heated to boiling. The boiling solution is poured in a stream through the separatory funnel into the flask upon the sodium and then washed down with 50 cc. of boiling isoamyl alcohol. A very violent reaction begins immediately which slows up after about five minutes and then must be aided by warming. The flame is so regulated that the reaction mixture is always boiling very vigorously until all the sodium is dissolved. This generally requires about three or four hours. If after three hours the sodium is not completely dissolved it is advisable, in order to hasten the reaction, to add 50 cc. more of isoamyl alcohol.
When all the sodium is completely dissolved, the hot, yellow solution is allowed to cool to about 100° and then poured into 500 cc. of cold water. The mixture is allowed to cool completely with frequent shaking, and then the upper isoamyl alcohol layer is separated from the water layer which contains the principal part of the sodium hydroxide.
The reduction is repeated four times with 20 g. of β-naphthylamine so that altogether 100 g. of naphthylamine has been reduced (Note 5). The isoamyl alcohol solutions from all five portions are combined, and concentrated hydrochloric acid is added until the solution is just acid to litmus. For this purpose usually about 40–50 cc. is needed (Note 6). The greater part of the isoamyl alcohol is now distilled from a large flask (about 900–1100 cc. is recovered). The distillation is continued until a crust of crystals starts to form, and the residue is then cooled and treated with 200–300 cc. of water and about 300 cc. of 50 per cent potassium hydroxide solution. The base which separates is extracted is the separatory funnel with four 400–500 cc. portions of ether and the ether extracts combined in a wide-necked flask.
The flask is set in ice water, and a rapid stream of carbon dioxide, which has previously passed through two wash bottles containing water (Note 7), is passed in. After about one-quarter hour the liquid begins to become cloudy, and soon after there follows a rather rapid separation of the carbonate of the strongly basic alicyclic compound; the aromatic base does not react but remains in solution. The carbondioxide is passed in continuously until no more carbonate separates; this requires about four to six hours. The carbonate is filtered by suction on a Büchner funnel and washed with a little dry ether. The filtrate is again treated with carbon dioxide; there is often obtained still more carbonate which is added to the precipitate (Note 8).
For further purification, the almost dry carbonate is dissolved in
500–600 cc. of 7 per cent acetic acid and the solution is filtered from the dark impurities. The clear, almost colorless solution is decomposed with a large excess of
potassium hydroxide (about 200 cc. of a 50 per cent solution), whereupon the alicyclic base separates as a light-brown oil which collects on the surface of the water solution and is extracted as rapidly as possible by shaking three or four times with
ether. The ether solution is dried for at least six hours with about
90–100 g. of potassium hydroxide (Note 9). The distillation of the
ether is carried out most conveniently in a specially designed
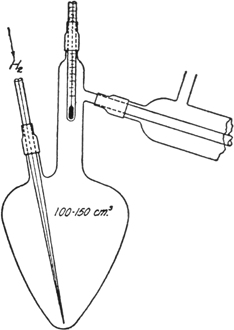
Claisen flask (Fig. 26) of 100–150 cc. capacity,
Fig. 26.
 |
the
ether solution gradually being added to the flask from the separatory funnel.
The base is distilled under the reduced pressure obtained by a water pump, because it is easily decomposed under ordinary pressure. For this distillation a flask of the form shown in Fig. 26 is recommended for use (Note 10) though it is not essential.
Since the base is sensitive to oxygen, dry hydrogen is passed through the capillary tube used in the distillation (Note 11). The distillation of the base runs very smoothly, and the liquid distils over from beginning to end almost constantly within a degree.
The yield of pure distilled base, b.p. 118.5°/8 mm., 127.5–128°/12 mm., 140–140.5°/20 mm., amounts to 53–59 g. (51–57 per cent of the theoretical amount). The pure base can be preserved only by sealing it in ampules (containing as little air as possible) immediately after distillation. The ac-tetrahydro-β-naphthylamine is a colorless, water-clear liquid which shows no fluorescence (Note 12) and possesses a strong odor similar to piperidine. In the air the base soon turns brown, rapidly absorbs carbon dioxide, and changes to the carbonate.
The hydrochloride of the ac-tetrahydro-β-naphthylamine can be obtained readily by neutralizing a dry ether solution of the base with an ether solution of hydrogen chloride. It crystallizes from water in large plates which melt at 237° (Note 13) and (Note 14).
2. Notes
1.
In place of the
sand bath it is possible to use an ordinary
wire gauze or a
Babo's air bath, but the danger of a fire due to a sudden breaking of the flask during the very violent reaction at the beginning is considerably greater. The experiment is best carried out using a sand bath. The hands should be protected with gloves and the eyes with glasses.
2.
After many experiments, this amount of
sodium was found to be the optimum. By the use of smaller quantities of
sodium and
β-naphthylamine the yield drops; with larger amounts the danger of breaking the glass flask increases. It is probably possible to use
copper flasks, but it is then impossible to see how the reaction is running.
3.
The stopcock of the separatory funnel must have as wide a hole as possible so that the
isoamyl alcohol can enter in a rapid stream.
4.
If a
water condenser is used the condensation is much more efficient but the danger of breaking is then much greater. A
brass inner tube in place of a glass one is desirable. If the condenser tube is too narrow, there is danger that the violently boiling
isoamyl alcohol will be thrown out at the beginning of the reaction because it cannot flow back.
5.
By working up less than
100 g. of β-naphthylamine the yield decreases rather rapidly on account of the various possibilities for loss in the later operations.
6.
An excess of acid does no harm but makes the recovery of the
isoamyl alcohol more difficult.
7.
If an
efficient reflux condenser is attached to the flask the major part of the
ether can be recovered. It is necessary to be sure that the
carbon dioxide is actually moist because otherwise the carbonate will not form at all or only very slowly.
8.
The
ether filtrates contain
dihydronaphthalene and ar-tetrahydro-β-naphthylamine.
9.
Since the free base reacts very strongly alkaline and also is sensitive, it is not possible to allow it to remain long in the air or loss will occur due to carbonate formation and oxidation.
10.
The suggested flask has the following advantages over other forms:
(a) The chance for contaminating the boiling base with rubber is reduced to a minimum; the capillary and thermometer are attached to the distillation flask by means of small rubber tubing.
(b) The loss due to the substance which remains on the walls of the flask is reduced to a minimum as compared to other flasks.
(c) The substances can, with the help of the capillary, be distilled over to the last drop, which is not possible in a round flask.
11.
This apparatus has also proved itself very useful for the distillation of other substances sensitive to
oxygen.
12.
Fluorescence arises from aromatic by-products which, however, can be entirely separated by careful work. Pure
ac-tetrahydro-β-naphthylamine gives no color with
diazobenzenesulfonic acid.
13.
According to Allewelt and Day, J. Org. Chem.
6, 386 (1941), the crude hydrochloride can be obtained in an almost colorless condition by shaking it with
150–200 cc. of ether in the cold.
14.
Of special interest is the extremely powerful physiological action of the tetrahydro base and its salts,
1 which makes it advisable to handle it carefully. The action can be called a symptom-complex to which Barger has assigned the name, "sympathomimetic," and it consists in a maximum dilation of the pupils (mydriasis), in a marked increase in the arterial blood pressure, and in an increase in the rapidity of breathing.
3. Discussion
ac-Tetrahydro-β-naphthylamine can be prepared by the reduction of
β-naphthylamine with
sodium and
alcohol, a method first described by Bamberger and Müller.
2 It is possible to substitute
ethyl alcohol for
amyl alcohol, but the yield is distinctly lower. The base has been obtained recently in good yield by hydrogenation of
β-naphthylamine using a
nickel catalyst.
3
Appendix
Chemical Abstracts Nomenclature (Collective Index Number);
(Registry Number)
ac-Tetrahydro-β-naphthylamine
hydrochloride of the ac-tetrahydro-β-naphthylamine
ar-tetrahydro-β-naphthylamine
ethyl alcohol,
alcohol (64-17-5)
hydrogen chloride,
hydrochloric acid (7647-01-0)
acetic acid (64-19-7)
ether (60-29-7)
hydrogen (1333-74-0)
sodium hydroxide (1310-73-2)
oxygen (7782-44-7)
carbon dioxide,
carbondioxide (124-38-9)
nickel (7440-02-0)
potassium hydroxide (1310-58-3)
sodium (13966-32-0)
piperidine (110-89-4)
2-Naphthylamine, 1,2,3,4-tetrahydro- (2954-50-9)
isoamyl alcohol (123-51-3)
naphthylamine (134-32-7)
dihydronaphthalene
diazobenzenesulfonic acid
amyl alcohol (71-41-0)
β-naphthylamine (91-59-8)
Copyright © 1921-, Organic Syntheses, Inc. All Rights Reserved