Org. Synth. 1940, 20, 1
DOI: 10.15227/orgsyn.020.0001
β-(3-ACENAPHTHOYL)PROPIONIC ACID
[Acenaphthenebutyric acid, γ-oxo]
Submitted by L. F. Fieser
Checked by W. W. Hartman and A. Weissberger.
1. Procedure
In a 3-l. round-bottomed three-necked flask (Note 1), 100 g. (0.65 mole) of pure acenaphthene (Note 2) and 72 g. (0.72 mole) of succinic anhydride are dissolved by warming in 600 ml. of nitrobenzene. The flask is clamped in a large ice bath. Through the central opening is inserted a mercury-sealed mechanical stirrer. A second opening is connected to a gas trap and also carries a thermometer; the third is for the introduction of aluminum chloride. After the mixture has been cooled to about 0°, 195 g. (1.46 moles) of aluminum chloride is added in small portions in the course of 1 hour, the temperature being kept below 5°. Stirring is continued at 0° for 4 more hours, after which time the mixture is allowed to stand for at least 12 hours so that the ice melts and the clear red solution gradually comes to room temperature.
The flask is cooled by immersion in a slush of ice and water, and the addition compound is decomposed by adding gradually 200 g. of ice, 100 ml. of water, and
100 ml. of concentrated hydrochloric acid (this is best done under a
hood). The keto acid separates in the form of a stiff, grayish white paste. The solvent is removed by steam distillation, in which operation it is advisable to use a very rapid flow of steam together with an efficient condensing system, such as that illustrated
(Fig. 1). The condensing flask shown need be no more than 1 l. in capacity, and the exit tube should be centered at the bottom of this flask so that it can drain the contents completely. Some condensate ordinarily remains in the flask to serve as a vapor seal, a factor which adds greatly to the efficiency of condensation. For purposes of inspection, the flask can be emptied by diverting for a moment the stream of water to one side of the flask. The stoppers which are under pressure should be secured with wire. The distilling flask is heated to prevent too much condensation of steam
(Note 3).
Fig. 1.
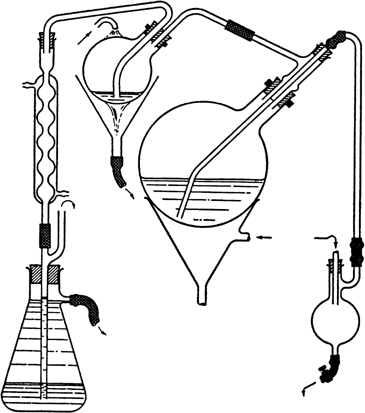
The bulk of the nitrobenzene comes over in about 1.5 hours, and the product then separates as a pasty mass which slowly disintegrates to a powder. During this process the elimination of nitrobenzene is very slow, but the steaming should be continued until only a few small lumps remain (4–5 hours), although it is not necessary to remove every trace of solvent (Note 4). The mixture is cooled with tap water, the crude acid is filtered and returned to the flask, and 115 g. of sodium carbonate decahydrate is added, together with sufficient water to make the flask a little less than half full. The mixture is heated with shaking over a free flame until most of the solid has dissolved and the frothing has diminished. A few drops of capryl alcohol may be added to dissipate the froth. The dark brown solution is steam-distilled to eliminate the last traces of nitrobenzene (about 30 minutes) and then filtered by suction from a very light residue. One hundred grams of sodium chloride is dissolved in the hot solution (volume, about 1.5 l.), which is then allowed to cool without disturbance. The sodium salt of β-(3-acenaphthoyl) propionic acid separates as colorless, fibrous needles, while the isomeric 1-acid largely remains in solution (Note 5). The product is collected on a large Büchner funnel and washed free of the dark mother liquor with half-saturated sodium chloride solution (about 150 ml.), the combined filtrates (A) being set aside. The sodium salt is crystallized once more from boiling water (1–1.5 l.) (Note 6), using Norit if required, and adding 50 g. of sodium chloride to the hot filtered solution. The mother liquor (B) is again saved. The purified salt is dissolved in 1.2 l. of hot water and the solution is acidified. The free acid separates as a white powder in a very pure condition. The yield of β-(3-acenaphthoyl) propionic acid melting at 206–208° with decomposition is 133 g. (81%) (Note 7).
2. Notes
1.
By using a flask suitable for steam distillation, the loss in time and material attending a transfer is avoided.
2.
Suitable material is supplied by Reilly Tar and Chemical Corporation, New York, or Gesellschaft für Teerverwertung, Duisburg-Meiderich, Germany.
3.
As compared with the apparatus shown in
Fig. 24 of Org. Syntheses Coll. Vol. 1, 479, this arrangement requires a much smaller flask and yet offers unlimited capacity. It also enables the operator to observe more closely the nature of the distillate.
4.
If the flask fills up with condensed steam, it should be cooled, the contents filtered, and the product returned to the flask. The process of disintegration can be hastened by breaking up the lumps with a flattened
stirring rod.
5.
The small amount of isomeric 1-acid may be obtained from the mother liquors, A and B. The first of these on acidification gives a product which is dark and tarry, but which soon solidifies on being cooled and stirred. The material is dissolved in 1 l. of water containing
25–30 g. of sodium carbonate decahydrate, and the solution is boiled for 30 minutes with Norit, filtered, cooled, and acidified. The product, which now solidifies at once and is lighter in color, is dried and combined with the material obtained by acidifying the second mother liquor, B (total amount, 23.9 g.). The crude mixture of acids is suspended in
170 ml. of cold methanol,
8.5 ml. of concentrated sulfuric acid is added, and the mixture is heated on the
steam bath for about 10 minutes, after which dissolution and esterification are complete. The dark product that crystallizes when the solution cools is largely the 1-ester, which is very much less soluble than the 3-ester. The 1-ester (
13.1 g.) is washed free of acid; it crystallizes from
ethanol with the use of decolorizing carbon in long needles melting at
126°; yield,
9 g. (Note 8). The ester is hydrolyzed by heating with
100 ml. of alcohol and
30 ml. of 25% sodium hydroxide solution until dissolved; the solution is then diluted with water and acidified. The
β-(1-acenaphthoyl)propionic acid melts at
180° (crystallized from dilute alcohol, 181°) and weighs
8.4 g. (
5%)
(Note 9).
6.
If, owing to hydrolysis, the sodium salt fails to dissolve completely, alkali should be added as required.
7.
The 3-acid crystallizes well from
glacial acetic acid, alcohol, or
xylene, but large volumes of solvent are required and there is no change in the melting point.
8.
The 3-ester melts at
89°.
9.
Ordinarily the mother liquors from the preparation and purification of 1-ester will be discarded, but a small additional quantity of the 3-acid may be obtained by concentrating these solutions, adding alkali to hydrolyze the ester, adding water, and acidifying. The precipitated material is purified by crystallizing the sodium salt twice, and from this
8 g. (
5%) of the pure 3-acid is obtained.
The ratio of 3-acid to 1-acid is dependent on the temperature, lower temperatures favoring the production of 3-acid. At −15° the yield of 3-acid is 87%, and of 1-acid, 5%. At room temperature there is some increase in the proportion of the 1-acid formed, but the product is very dark and difficult to work up, and the total yield is lower even though the aluminum chloride is added in nitrobenzene solution.
3. Discussion
This procedure is based upon a study
1 of the method outlined in the patent literature.
2 The procedure is a general one and may be used for the condensation of
succinic anhydride with
naphthalene and with the mono- and dimethylnaphthalenes, although in no other case are the purification and separation of isomers so easily accomplished. In this particular type of condensation, as well as in certain other types of Friedel-Crafts reactions,
nitrobenzene is far superior to the solvents that are more frequently employed. This is partly because of its great solvent power and partly because it forms a molecular compound with
aluminum chloride, and so decreases the activity of the catalyst in promoting side reactions.
Appendix
Chemical Abstracts Nomenclature (Collective Index Number);
(Registry Number)
alcohol,
ethanol (64-17-5)
sulfuric acid (7664-93-9)
hydrochloric acid (7647-01-0)
acetic acid (64-19-7)
methanol (67-56-1)
sodium hydroxide (1310-73-2)
sodium chloride (7647-14-5)
Norit (7782-42-5)
aluminum chloride (3495-54-3)
Nitrobenzene (98-95-3)
Naphthalene (91-20-3)
sodium carbonate decahydrate (6132-02-1)
xylene (106-42-3)
Succinic anhydride (108-30-5)
capryl alcohol (111-87-5)
acenaphthene (83-32-9)
β-(3-Acenaphthoyl)propionic acid,
β-(3-acenaphthoyl) propionic acid (16294-60-3)
Acenaphthenebutyric acid, γ-oxo,
β-(1-acenaphthoyl)propionic acid
sodium salt of β-(3-acenaphthoyl) propionic acid
Copyright © 1921-, Organic Syntheses, Inc. All Rights Reserved