Org. Synth. 1967, 47, 4
DOI: 10.15227/orgsyn.047.0004
1,2,3-BENZOTHIADIAZOLE 1,1-DIOXIDE
Submitted by G. Wittig and R. W. Hoffmann
1.
Checked by C. D. Smith, R. A. Clement, and B. C. McKusick.
1. Procedure
A. 2-Nitrobenzenesulfinic acid (Note 1). Caution! This reaction should be done in a good hood because noxious fumes are released.
2-Nitroaniline (13.8 g., 0.10 mole) (Note 2) is dissolved in a hot solution of 75 ml. of 96% sulfuric acid, 100 ml. of phosphoric acid (density 1.7), and 50 ml. of water in a 1-l. beaker. A stirrer and a thermometer are introduced into the mixture, and the beaker is immersed in an ice bath. A solution of 8.3 g. (0.12 mole) of sodium nitrite in 25 ml. of water is added dropwise to the well-stirred solution at such a rate that the temperature is maintained at 10–15°. Excess nitrite is destroyed by adding sulfamic acid in small portions (Note 3). The mixture is cooled to −10° in an ice-salt bath, and about 50 ml. of liquid sulfur dioxide (Note 4) is poured into the well-stirred reaction. The product is immediately poured onto a mixture of 55.6 g. (0.20 mole) of FeSO4·7 H2O and 1 g. of defatted copper powder in a wide 2-l. beaker. Nitrogen and excess sulfur dioxide bubble off with much foaming.
After 30 minutes the solid sulfinic acid is separated on a fritted-glass filter. The sulfinic acid is dissolved from the filter by a mixture of 750 ml. of ether and 750 ml. of methylene chloride. The solution is dried over calcium chloride and evaporated to dryness under reduced pressure (bath temperature 25°) (Note 5). The residue is suspended in 50 ml. of water, and small portions of dilute ammonia are added to the well-stirred suspension until it has a pH of 9 (Note 6). Insoluble impurities are separated by filtration, and 2-nitrobenzenesulfinic acid is precipitated from the filtrate by adding 5-ml. portions of 6N hydrochloric acid with cooling; the sulfinic acid precipitated by each portion of acid is separately collected on a Buchner funnel (Note 7). The acid, a pale yellow solid, is dried on a clay plate in a vacuum desiccator over potassium hydroxide pellets, m.p. 120–125° (dec.), weight 9.4–14.9 g. (50–80%). If the 2-nitrobenzenesulfinic acid is to be used for the hydrogenation of the next step high purity is required, and it is generally advisable to reprecipitate the acid once more in the same way (Note 8).
B.
Sodium 2-aminobenzenesulfinate.
2-Nitrobenzenesulfinic acid (3.74 g., 0.020 mole) is suspended in 10 ml. of water, and sufficient
1N NaOH (about 20 ml.) is added to the well-stirred mixture to dissolve the acid and bring the pH to 9.
Palladium oxide (0.2–1.0 g., (Note 9)) is suspended in 20 ml. of water in a
200-ml. glass hydrogenation bottle. The bottle is attached to a hydrogenation apparatus such as that of Adams and Voorhees,
2 and the suspension is shaken with
hydrogen under a pressure of 1–3 atm. until the
palladium oxide is reduced. The solution of
2-nitrobenzenesulfinic acid is added, and the mixture is shaken under a
hydrogen pressure of 1–3 atm. The solution becomes completely decolorized in 2–6 hours, during which time about
95% of the calculated amount of hydrogen is absorbed. The catalyst is separated by filtration and washed with two 20-ml. portions of water, which are added to the filtrate. The filtrate, which may have a yellowish color, is evaporated to dryness under reduced pressure (bath temperature 45°). The residue, a white or light yellow solid, is
sodium 2-aminobenzenesulfinate. After being dried in a
desiccator over
calcium chloride, it weighs
3.05–3.20 g. (
85–89%).
C. 1,2,3-Benzothiadiazole 1,1-dioxide. Caution! 1,2,3-Benzothiadiazole 1,1-dioxide in the solid state can explode spontaneously, particularly on being warmed, jolted, or scratched. For most purposes it need not be isolated, but can be used in solutions, which are relatively safe. Any operations involving the solid material should be done very carefully, using good shielding.
A solution of 1.43 g. (0.0080 mole) of sodium 2-aminobenzenesulfinate in the least possible amount of water is combined with a solution of 0.55 g. (0.0080 mole) of sodium nitrite in the least amount of water. A mixture of 16 ml. of 2N sulfuric acid and 22 ml. of glycerol is placed in a 250-ml. three-necked flask equipped with a dropping funnel, a low-temperature thermometer, and a stirrer, and the flask is immersed in a bath of acetone and dry ice. The stirred mixture is cooled to −15°, and the solution of sodium 2-aminobenzenesulfinate and sodium nitrite is added dropwise over a period of about 5 minutes; the cooling and rate of addition are such as to maintain the temperature at −15° ± 3°. The mixture is stirred for an additional 2 hours at this temperature, and 30 ml. of ether is added. The product is stirred vigorously for a few minutes and then allowed to warm to −6° with gentle stirring. The ether layer is decanted or transferred by means of a chilled pipet into a vessel cooled in a dry ice bath, and the reaction mixture is again cooled to −15°. In this way the reaction mixture is extracted with five 20-ml. portions of ether. After the last extraction the aqueous layer is frozen solid and the ether layer is poured off. The combined extracts are dried at −20°, first over calcium chloride and then over phosphorus pentoxide; a cold room at −20° is particularly convenient for this operation. The solution is transferred to a tared distillation flask immersed in an ice bath (Note 10), and the ether is removed by evaporation under reduced pressure. The flask is weighed rapidly and dried in a desiccator over phosphorus pentoxide at −20° (
Caution! (Note 11)(Note 12)). The residue is 1,2,3-benzothiadiazole 1,1-dioxide in the form of yellow-brown needles; weight 0.77–1.04 g. (57–77%). It explodes between 45° and 60° (Note 13).
2. Notes
1.
This is essentially the method of J. Lange.
32.
Technical material of Badische Anilin & Soda-Fabrik is satisfactory.
3.
To detect
nitrous acid, a drop of the mixture is diluted with water and tested with starch iodide paper.
4.
It is convenient to condense
sulfur dioxide from a
cylinder in a calibrated trap cooled in a dry ice bath.
5.
After the procedure had been checked, the submitters recommended the following time-saving modification. The
methylene chloride-
ether solution of the sulfinic acid is extracted with one
80-ml. portion and two 35-ml. portions of 2N sodium hydroxide solution. The extracts are combined and the sulfinic acid is precipitated with
5-ml. portions of 6N hydrochloric acid as described in the text.
6.
An excess of
ammonia leads to products that are contaminated with
ammonium chloride.
7.
In this way one avoids an excess of
hydrochloric acid which, if it adheres to the product, causes its gradual decomposition.
8.
The checkers dissolved the crude acid in the minimum amount of
2N sodium hydroxide (about 3 ml./g.) and reprecipitated it in 5 portions with 2
N hydrochloric acid; recovery
75–85%. Alternatively, they added the acid to boiling
ethyl acetate (9 ml./g.), added
decolorizing carbon to the solution, boiled the mixture for 5 minutes, separated the carbon by filtration, and cooled the hot filtrate; recovery
45–55%. The checkers found no difference in the infrared spectra of material purified in the two ways, but recrystallized material was reduced more quickly by
hydrogen.
9.
The submitters used
0.2 g. of palladium oxide prepared by the method of Shriner and Adams
4 and required 2 hours for complete hydrogenation under a
hydrogen pressure of 1 atm. The checkers used
1.0 g. of palladium oxide (75.7%) from Engelhard Industries, 113 Astor Street, Newark, New Jersey, and required 4 hours for complete hydrogenation under a
hydrogen pressure of 2–3 atm. Conditions should be chosen to give complete hydrogenation within 6 hours or colored by-products may be formed.
10.
Not quite half of the flask should dip into the ice water or the layer of ice forming on the flask may be hard to remove.
11.
1,2,3-Benzothiadiazole 1,1-dioxide slowly decomposes even at 0°; hence it should always be used on the day on which it is made. For most purposes it is not necessary to isolate the dioxide; the
ether solution can be used, or solutions in other solvents can be prepared by adding the other solvent and distilling off the
ether under reduced pressure (bath temperature 0°). In this way larger amounts of the dioxide than are described in this procedure can be handled without danger.
12.
1,2,3-Benzothiadiazole 1,1-dioxide can be conveniently assayed and characterized without isolation by forming its adduct with
cyclopentadiene.
5 The following procedure illustrates characterization; for assay the same procedure can be applied to an aliquot, with all amounts scaled down in proportion. The dried
ether extract of
1,2,3-benzothiadiazole 1,1-dioxide prepared from
1.43 g. (0.0080 mole) of sodium 2-aminobenzenesulfinate is concentrated to about 20 ml. at 0°, and
20 ml. of acetonitrile at −20° is added.
Twenty milliliters of cold, freshly prepared cyclopentadiene6 is added. The mixture is kept overnight at −10° to 0°. Solvent and excess
cyclopentadiene are removed by evaporation at 0° under reduced pressure to leave 1.20–1.28 g. (64–68% based on
sodium 2-aminobenzenesulfinate) of crude 1:1 adduct, m.p. 87° (dec.). For purification it is dissolved in
20 ml. of methylene chloride,
70 ml. of ether is added, and the solution is kept at −70°. Adduct decomposing at 90° crystallizes; recovery is about 75%. From pure, crystalline
1,2,3-benzothiadiazole 1,1-dioxide the yield of adduct is 92–98%.
13.
Purer product can be obtained by reducing
1,2,3-benzothiadiazole 1,1-dioxide with
zinc and acetic acid to
1,2,3-benzothiadiazoline 1,1-dioxide, which is oxidized back with
lead tetraacetate.
5
3. Discussion
1,2,3-Benzothiadiazole 1,1-dioxide has been prepared only by the present method.
5
4. Merits of the Preparation
1,2,3-Benzothiadiazole 1,1-dioxide decomposes smoothly in solution at 10° to give
dehydrobenzene ("benzyne"),
nitrogen, and
sulfur dioxide.
5,7 In this way, as well as by the thermal decomposition of
benzenediazonium-2-carboxylate,
8,9 it is possible to obtain
dehydrobenzene in the absence of organometallic or strongly alkaline reagents; for this reason the choice of the reaction partner for
dehydrobenzene is hardly limited at all. Compared to dry
benzenediazonium-2-carboxylate,
1,2,3-benzothiadiazole 1,1-dioxide possesses the following advantages as a source of
dehydrobenzene: the explosive compound does not need to be isolated and the decomposition temperature is lower. Solvent-wet
benzenediazonium-2-carboxylate, being insoluble in most organic media, is less generally useful than
1,2,3-benzothiadiazole 1,1-dioxide but is more convenient to prepare.
9 Because of their special reaction conditions, other methods of obtaining
dehydrobenzene without using an organometallic compound
10 are not so generally applicable. Earlier volumes of
Organic Syntheses illustrate the preparation of
dehydrobenzene by the action of
magnesium on
o-fluorobromobenzene11 and a type of ring closure in which a
dehydrobenzene is an intermediate.
12 Methods of generating dehydrobenzenes and the reactions of these reactive substances were recently reviewed.
13
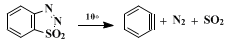
Appendix
Chemical Abstracts Nomenclature (Collective Index Number);
(Registry Number)
H2O
dehydrobenzene
calcium chloride (10043-52-4)
sulfuric acid (7664-93-9)
hydrochloric acid (7647-01-0)
acetic acid (64-19-7)
ammonia (7664-41-7)
ethyl acetate (141-78-6)
ether (60-29-7)
ammonium chloride (12125-02-9)
hydrogen (1333-74-0)
acetonitrile (75-05-8)
sodium hydroxide (1310-73-2)
glycerol (56-81-5)
magnesium (7439-95-4)
sulfur dioxide (7446-09-5)
nitrogen (7727-37-9)
sodium nitrite (7632-00-0)
nitrous acid (7782-77-6)
copper powder (7440-50-8)
potassium hydroxide (1310-58-3)
zinc (7440-66-6)
phosphoric acid (7664-38-2)
methylene chloride (75-09-2)
palladium oxide
sulfamic acid (5329-14-6)
CYCLOPENTADIENE (542-92-7)
Benzenediazonium-2-carboxylate (1608-42-0)
1,2,3-Benzothiadiazole 1,1-dioxide (37150-27-9)
2-Nitrobenzenesulfinic acid (13165-79-2)
2-Nitroaniline (88-74-4)
Sodium 2-aminobenzenesulfinate (50827-53-7)
1,2,3-benzothiadiazoline 1,1-dioxide
o-fluorobromobenzene (1072-85-1)
phosphorus pentoxide (1314-56-3)
lead tetraacetate (546-67-8)
Copyright © 1921-, Organic Syntheses, Inc. All Rights Reserved