Org. Synth. 1979, 59, 169
DOI: 10.15227/orgsyn.059.0169
STEREOSELECTIVE HYDROXYLATION WITH THALLIUM(I) ACETATE AND IODINE: trans- AND cis-1,2-CYCLOHEXANEDIOLS
Submitted by R. C. Cambie and P. S. Rutledge
1.
Checked by D. Seebach, M. Liesner, and E.-M. Wilka.
1. Procedure
Caution! Thallium salts are very toxic. These procedures should be carried out in a
well-ventilated hood, and rubber gloves should be worn. For disposal of thallium wastes, see
Org. Synth., Coll. Vol. 6, 791 (1988).
A. trans-1,2-Cyclohexanediol. In a 100-ml., round-bottomed flask equipped with a reflux condenser protected with a drying tube are placed a magnetic stirring bar, 17.56 g. (0.05447 mole) of thallium(I) acetate (Note 1), and 40 ml. of dried acetic acid (Note 2). The mixture is stirred and heated at reflux for 1 hour. The mixture is cooled before 2.84 g. (3.50 ml., 0.0346 mole) of cyclohexene (Note 3) and 8.46 g. (0.0333 mole) of iodine (Note 4) are added. The resulting suspension is stirred and heated at reflux for 9 hours (Note 5), and then cooled to room temperature. The yellow thallium(I) iodide precipitate is filtered and washed thoroughly with diethyl ether. The filtrates are combined, the solvents are removed with a rotary evaporator (Note 6), and the residual liquid is dissolved in dry ether. The turbid solution is dried with anhydrous potassium carbonate, and the solvent is again removed by rotary evaporation (Note 6), affording 5.4–6.3 g. of trans-1,2-cyclohexanediol diacetate as a mobile, brown liquid (Note 7).
The diacetate is dissolved in 25 ml. of 95% ethanol, a solution of 2.9 g. (0.073 mole) of sodium hydroxide in 11 ml. of water is added, and the resulting mixture is heated at reflux for 3 hours. The solution is concentrated by rotary evaporation and the remaining syrup is extracted with six 50-ml. portions of chloroform. The combined extracts are dried over anhydrous magnesium sulfate and evaporated, providing 3.1–3.3 g. of a pale brown crystalline solid, m.p. 97–103°. Recrystallization from carbon tetrachloride gives 2.5–2.7 g. (65–70% based on iodine) of trans-1,2-cyclohexanediol, m.p. 103–104° (Note 8).
B. cis-1,2-Cyclohexanediol. A 500-ml., round-bottomed flask equipped with a reflux condenser and a magnetic stirring bar is charged with 17.56 g. (0.05447 mole) of thallium(I) acetate(Note 1), 160 ml. of glacial acetic acid, 3.0 g. (3.7 ml., 0.036 mole) of cyclohexene (Note 3), and 8.46 g. (0.0333 mole) of iodine (Note 4) in the order given. The suspension is stirred and warmed in a heating bath at 80° for 30 minutes. An 80-ml. portion of water is added, stirring is continued, and the mixture is heated at reflux for 9 hours. The product is isolated and hydrolyzed as described in Part A, affording 3.2–4.9 g. of cis-1,2-cyclohexanediol, m.p. 88–95° (Note 9). Recrystallization from carbon tetrachloride gives 2.7–2.9 g. (70–75% based on iodine) of cis-1,2-cyclohexanediol, m.p. 97–98° (Note 10).
2. Notes
1.
Thallium(I) acetate purchased from BDH Chemicals Ltd., Poole, England, or Fluka AG, Buchs, Switzerland, was used without further purification. This reagent is also available from Alfa Division, Ventron Corporation.
2.
The submitters purchased
glacial acetic acid from Showa Denko K. K., Tokyo, Japan, and
acetic anhydride from Riedel de Haen AG, Seelze-Hannover, Germany. A solution prepared from
4 volumes of glacial acetic acid,
1 volume of acetic anhydride, and a catalytic amount of
p-toluenesulfonic acid was heated under reflux for 24 hours and distilled. The distillate, which contained 5% water and
4% acetic anhydride according to
1H NMR, analysis, was then used by the submitters. The water content was determined from the chemical shift of the hydroxyl proton.
2The checkers purchased analytical grade glacial acetic acid and anhydride from E. Merck, Darmstadt, Germany. A 4:1 (v/v) solution of glacial acetic acid and acetic anhydride containing 500 mg. of p-toluenesulfonic acid per liter was heated at reflux for 24 hours and then distilled. A forerun corresponding to 25% of the solution was discarded before a main fraction of acetic acid corresponding to 60% of the solution was collected. The main fraction, containing 17% acetic anhydride and 1% or less water as determined from its 1H NMR spectrum, was used by the checkers.
3.
Cyclohexene was purchased from BDH Chemicals Ltd., Poole, England, by the submitters and used without purification. This reagent was purchased by the checkers from Fluka AG, Buchs, Switzerland, and distilled before use.
4.
Iodine purchased from Riedel de Haen AG, Seelze-Hannover, Germany, was sublimed before use by the submitters. The checkers used
iodine from Siegfried AG, Zofingen, Switzerland, without purification.
5.
After
ca. 30 minutes the initially black-green solid becomes yellow.
6.
The checkers recommend that excessive heating and evacuation be avoided during rotary evaporation to minimize the loss of product during this operation. They kept the heating bath temperature below 80° and used a
water aspirator.
7.
The diacetate was judged to be virtually pure by the submitters on the basis of GC analysis carried out at 150° using a
glass column packed with 3% OV 17 (
1:1 methyl–phenyl silicone) supported on 70–80 mesh Chromosorb W.
8.
The submitters obtained
2.9 g. (
75%) of product that melted at
103–105° (lit.,
3 m.p.
104°).
9.
The unrecrystallized product obtained by the submitters melted at
91–95°. The purity of this material is estimated to be 96% on the basis of the melting point.
310.
The submitters obtained
3.0 g. (
78%) of product which melted at
99–100°. The melting point of
97–98° shown above was obtained by the checkers using a
Tottoli melting point apparatus (Büchi) equipped with a 50° range Anschütz thermometer. A melting point of
98° has been reported.
4
3. Discussion
The present procedure offers a convenient alternative to the Prévost reaction
5 6 and the Woodward modification of the Prévost reaction
7 in which silver carboxylates are used instead of thallium(I) carboxylates. Thallium(I) salts have the advantages of being generally stable crystalline solids that can be readily prepared in high yield by neutralization of the appropriate carboxylic acid with
thallium(I) ethoxide.
8 Silver salts, on the other hand, are frequently unstable and difficult to dry.
Thallium and its compounds are, however, extremely toxic, and great care must be taken in the use and disposal of thallium salts.
9,10,11The mechanisms of these reactions are presumably analogous to those of the Prévost and Woodward–Prévost reactions.
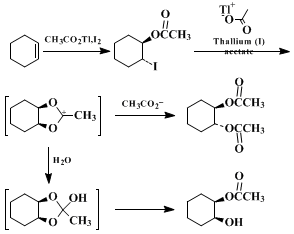
5,6,7 In the first step of the reaction of
iodine and
thallium(I) acetate with
cyclohexene, in both parts A and B of this procedure, produces
trans-2-odocyclohexyl acetate. The second equivalent of
thallium(I) acetate scavenges iodide ion during formation of the 1,3-dioxolan-2-ylium ion intermediate. Under the anhydrous conditions in Part A, the carbonium ion reacts with acetate ion at a ring carbon with inversion to give the diacetate. In part B the ion is captured by water, and the resulting ortho ester undergoes ring opening to the
cis-diol monoacetate. No appreciable reaction occurs unless
thallium(I) acetate,
iodine, and
cyclohexene are all present. Thus, in contrast to the Prévost and Woodward–Prévost procedures,
acetyl hypoiodite evidently cannot be prepared separately from
thallium(I) acetate and
iodine. The precise reasons for this difference are not clear.
trans-2-Iodocyclohexyl acetate can be isolated in essentially quantitative yield from the reaction of
thallium(I) acetate,
iodine, and
cyclohexene in a 1:1:1 molar ratio in refluxing
chloroform.
12 Similarly, iodo acetates from a representative series of alkenes, including
cyclohexene, have been prepared in 80–98% yield
13 in glacial
acetic acid which was not dried as described in this procedure. The corresponding iodo benzoates are obtained in comparable yields from reaction with
thallium(I) benzoate and
iodine in
benzene. The deactivated olefin
methyl cinnamate did not react under these conditions, and
o-allylphenol underwent ring iodination to give
2-allyl-6-iodophenol.
14 The diterpenes,
phyllocladene and
isophyllocladene, upon reaction with
thallium(I) benzoate and
iodine,
15 afford the same mixture of allylic benzoates obtained from a Woodward–Prévost reaction. With the exception of
3-phenylpropene, the formation of iodo carboxylates from unsymmetrical alkenes occurs regioselectively in a Markovnikov sense.
Vicinal iodo carboxylates may also be prepared from the reaction of olefins either with
iodine and
potassium iodate in
acetic acid,
16 or with
N-iodosuccinimide and a carboxylic acid in
chloroform.
17 A number of new procedures for effecting the hydroxylation or acyloxylation of olefins in a manner similar to the Prévost or Woodward–Prévost reactions include the following: iodo acetoxylation with
iodine and
potassium chlorate in
acetic acid followed by acetolysis with
potassium acetate;
18 reaction with
N-bromoacetamide and
silver acetate in
acetic acid;
22 reaction with
thallium(III) acetate in
acetic acid;
19 and reaction with
iodine tris(trifluoroacetate) in
pentane.
20The preparation of
trans-1,2-cyclohexanediol by oxidation of
cyclohexene with
performic acid and subsequent hydrolysis of the diol monoformate has been described,
21,10,11The mechanisms of these reactions are presumably analogous to those of the Prévost and Woodward–Prévost reactions.
5,6,7 In the first step of the reaction of
iodine and
thallium(I) acetate with
cyclohexene, in both parts A and B of this procedure, produces
trans-2-odocyclohexyl acetate. The second equivalent of
thallium(I) acetate scavenges iodide ion during formation of the 1,3-dioxolan-2-ylium ion intermediate. Under the anhydrous conditions in Part A, the carbonium ion reacts with acetate ion at a ring carbon with inversion to give the diacetate. In part B the ion is captured by water, and the resulting ortho ester undergoes ring opening to the
cis-diol monoacetate. No appreciable reaction occurs unless
thallium(I) acetate,
iodine, and
cyclohexene are all present. Thus, in contrast to the Prévost and Woodward–Prévost procedures,
acetyl hypoiodite evidently cannot be prepared separately from
thallium(I) acetate and
iodine. The precise reasons for this difference are not clear.
trans-2-Iodocyclohexyl acetate can be isolated in essentially quantitative yield from the reaction of
thallium(I) acetate,
iodine, and
cyclohexene in a 1:1:1 molar ratio in refluxing
chloroform.
12 Similarly, iodo acetates from a representative series of alkenes, including
cyclohexene, have been prepared in 80–98% yield
13 in glacial
acetic acid which was not dried as described in this procedure. The corresponding iodo benzoates are obtained in comparable yields from reaction with
thallium(I) benzoate and
iodine in
benzene. The deactivated olefin
methyl cinnamate did not react under these conditions, and
o-allylphenol underwent ring iodination to give
2-allyl-6-iodophenol.
14 The diterpenes,
phyllocladene and
isophyllocladene, upon reaction with
thallium(I) benzoate and
iodine,
15 afford the same mixture of allylic benzoates obtained from a Woodward–Prévost reaction. With the exception of
3-phenylpropene, the formation of iodo carboxylates from unsymmetrical alkenes occurs regioselectively in a Markovnikov sense.
Vicinal iodo carboxylates may also be prepared from the reaction of olefins either with
iodine and
potassium iodate in
acetic acid,
16,17,18 A number of new procedures for effecting the hydroxylation or acyloxylation of olefins in a manner similar to the Prévost or Woodward–Prévost reactions include the following: iodo acetoxylation with
iodine and
potassium chlorate in
acetic acid followed by acetolysis with
potassium acetate;
18 reaction with
N-bromoacetamide and
silver acetate in
acetic acid;
22 reaction with
thallium(III) acetate in
acetic acid;
19 and reaction with
iodine tris(trifluoroacetate) in
pentane.
20The preparation of
trans-1,2-cyclohexanediol by oxidation of
cyclohexene with
performic acid and subsequent hydrolysis of the diol monoformate has been described,
21 and other methods for the preparation of both
cis- and
trans-1,2-cyclohexanediols have been cited. Subsequently the
trans diol has been prepared by oxidation of
cyclohexene with various peracids,
23 with
hydrogen peroxide and
selenium dioxide,
24 and with
iodine and
silver acetate by the Prévost reaction.
25 Alternative methods for preparing the
trans isomer are hydroboration of various enol derivatives of
cyclohexanone26 and reduction of
trans-2-cyclohexen-1-ol epoxide with
lithium aluminum hydride.
27 cis-1,2-Cyclohexanediol has been prepared by
cis hydroxylation of
cyclohexene with various reagents or catalysts derived from
osmium tetroxide,
28 by solvolysis of
trans-2-halocyclohexanol esters in a manner similar to that of the Woodward–Prévost reaction,
18,22,20,25,29 by reduction of
cis-2-cyclohexen-1-ol epoxide with
lithium aluminum hydride,
27 and by oxymercuration of
2-cyclohexen-1-ol with
mercury(II) trifluoroacetate in the presence of
chloral and subsequent reduction.
30
Appendix
Chemical Abstracts Nomenclature (Collective Index Number);
(Registry Number)
methyl silicone
acetic acid and anhydride
performic acid
phyllocladene
isophyllocladene
phenyl silicone
ethanol (64-17-5)
potassium carbonate (584-08-7)
acetic acid (64-19-7)
Benzene (71-43-2)
ether,
diethyl ether (60-29-7)
acetic anhydride (108-24-7)
sodium hydroxide (1310-73-2)
chloroform (67-66-3)
Cyclohexanone (108-94-1)
Cyclohexene (110-83-8)
carbon tetrachloride (56-23-5)
iodine (7553-56-2)
selenium dioxide (7446-08-4)
potassium chlorate (3811-04-9)
hydrogen peroxide (7722-84-1)
Pentane (109-66-0)
magnesium sulfate (7487-88-9)
chloral (75-87-6)
potassium acetate (127-08-2)
silver acetate (563-63-3)
lithium aluminum hydride (16853-85-3)
osmium tetroxide (20816-12-0)
N-Bromoacetamide (79-15-2)
3-phenylpropene (300-57-2)
2-cyclohexen-1-ol
N-Iodosuccinimide (516-12-1)
p-toluenesulfonic acid (104-15-4)
THALLIUM(I) ACETATE (563-68-8)
Thallium (7440-28-0)
thallium(I) iodide
thallium(I) ethoxide (20398-06-5)
acetyl hypoiodite
thallium(I) benzoate
methyl cinnamate (103-26-4)
2-allyl-6-iodophenol
potassium iodate (7758-05-6)
thallium(III) acetate (2570-63-0)
iodine tris(trifluoroacetate)
mercury(II) trifluoroacetate (13257-51-7)
trans-1,2-Cyclohexanediol (1460-57-7)
cis-1,2-Cyclohexanediol (1792-81-0)
trans-1,2-cyclohexanediol diacetate
trans-2-iodocyclohexyl acetate
o-allylphenol (1745-81-9)
trans-2-cyclohexen-1-ol epoxide
cis-2-cyclohexen-1-ol epoxide
trans-2-odocyclohexyl acetate
Copyright © 1921-, Organic Syntheses, Inc. All Rights Reserved