Org. Synth. 1974, 54, 88
DOI: 10.15227/orgsyn.054.0088
MACROCYCLIC DIIMINES: 1,10-DIAZACYCLOÖCTADECANE
Submitted by Chung Ho Park and Howard E. Simmons
1.
Checked by Stephen R. Wilson and Robert E. Ireland.
1. Procedure
Caution! Benzene has been identified as a carcinogen; OSHA has issued emergency standards on its use. All procedures involving benzene should be carried out in a well-ventilated hood, and glove protection is required.
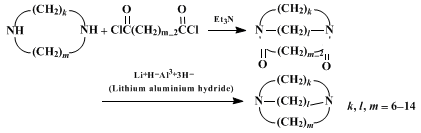
A. 1,10-Diazacycloöctadecane-2,9-dione. A 12-l., 4-necked, round-bottomed flask with four indents is fitted with a mechanical stirrer, two dropping funnels (Note 1), and an inlet tube to maintain a static nitrogen atmosphere throughout the reaction. Four liters of benzene (Note 2) are placed in the flask and stirred vigorously. Two solutions, one of 33.8 g. (0.160 mole) of suberyl dichloride (Note 3) in 2.0 l. of benzene and the other of 48.2 g. (0.335 mole) of 1,8-diaminoöctane (Note 4) in 2.0 l. of benzene, are added simultaneously over a 6–7 hour period at room temperature. After the addition is complete, the resulting suspension is stirred slowly overnight. The addition funnels are removed from the flask and replaced by stoppers fitted with tubes of suitable dimensions to permit the reaction mixture to be siphoned from the reaction flask when a slight positive nitrogen pressure is present in the flask. The fine suspension in the reaction flask is agitated and siphoned into a large, fritted, filter funnel. The white solid is washed three times with benzene and dried in a vacuum oven. The resulting white solid is pulverized, placed in a continuous extractor (Note 5), and extracted for three days with 1 l. of boiling benzene in a 2-l., round-bottomed flask equipped with a magnetic stirring bar and heating mantle. After the extractor is allowed to cool to room temperature, filtration of the white solid suspension gives 23.1–23.6 g. (51–52%) of crude 1,10-diazacycloöctadecane-2,9-dione, m.p. 198–201°, which is used in the subsequent step without further purification (Note 6).
B. 1,10-Diazacycloöctadecane. A 500-ml., round-bottomed flask equipped with a mechanical stirrer, condenser, and nitrogen bubbler is charged with 150 ml. of dry tetrahydrofuran (Note 7) and 3.8 g. (0.10 mole; 33% excess) of lithium aluminum hydride. While the suspension is stirred, 14.1 g. (0.0500 mole) of 1,10-diazacycloöctadecane-2,9-dione is added in small portions through a Gooch tube (Note 8). When the addition is complete and evolution of hydrogen subsides, the mixture is heated at reflux under a nitrogen atmosphere for 48 hours. The mixture is cooled to 5–10° with an ice bath and decomposed by cautious, dropwise addition of 3.8 ml. of water, followed by 3.8 ml. of 15% sodium hydroxide, and finally by 11.8 ml. of water. The mixture is allowed to come to room temperature, stirred for an additional hour, and filtered through a fritted-glass funnel. The resulting cake is washed thoroughly with three 50-ml. portions of tetrahydrofuran, followed by three 50-ml. portions of diethyl ether. The combined filtrate is concentrated under reduced pressure. After purging with benzene to remove traces of water and storing under vacuum overnight, 12.7 g. of crude diimine remains as a colorless, waxy solid.
The crude product contains a small amount of unreduced dilactam, which can be detected in the IR spectrum. Treatment of the crude diimine again under the identical reduction conditions (Note 9) using the same amounts of the reagent and solvent gives 10.6 g. (83%) of product. Distillation under reduced pressure through a semimicro Vigreux column (10-cm.) yields 9.5 g. (75%) of pure 1,10-diazacycloöctadecane, b.p. 120–130° (0.05 mm.), m.p. 59–61° (Note 10).
2. Notes
1.
Two graduated,
500-ml., pressure-equalizing dropping funnels with small diameter tips to allow solutions to be added in fine streams were used. They were equipped with screw-in type plungers with Teflon®
fluorocarbon tips to allow fine adjustment of the flow rates. The checkers utilized standard,
500-ml. dropping funnels and two needle valve adapters, as shown in
Figure 1. It is important that the two solutions are added at the same slow rate. The submitters report that mixing of the two solutions in one portion halved the yield of the dilactam.
Figure 1.
2.
All of the
benzene used was dried over
sodium wire and distilled from
sodium under a
nitrogen atmosphere.
3.
Suberyl dichloride purchased from Frinton Laboratories was used without further purification. The purity of the acid chloride was checked by GC after converting it to the diethyl ester.
4.
1,8-Diaminoöctane was purchased from Columbia Organic Chemicals Co., Inc. The purity was checked by GC and varied from batch to batch; only samples which were shown to be homogeneous were used. The diamine should be handled in an inert atmosphere, as it forms a carbonate rapidly.
5.
A
2-l. extractor with a fritted disc with an 85 mm. diameter and coarse porosity was used (
Figure 2).
Figure 2.
The desired product is only slightly soluble in benzene. Depending on the efficiency of extraction, this step may be extended to longer periods with beneficial results. A lower yield of the dilactam generally indicates incomplete extraction. An attempt to isolate the product by washing away the dihydrochloride from the solid mixture with water resulted in the formation of an emulsion.
6.
A sample of the crude dilactam dissolved in an
ethanol–water mixture gave no precipitate with
silver nitrate, indicating that the product is not contaminated with
octamethylenediamine dihydrochloride. If the purity is questionable, the crude product can be conveniently purified at this point by recrystallization from
N,N-dimethylformamide or
50% ethanol in water, giving better than
90% recovery of pure product, m.p.
207–209°.
7.
Commercially available
tetrahydrofuran was distilled from LiAlH
4 prior to use.
8.
The submitters suggest that on ten times this scale, the reaction flask be purged with a stream of
nitrogen during the addition of amide.
9.
The repeated treatment with
lithium aluminum hydride was necessary to obtain complete reduction, since use of the reducing agent in manyfold excess and longer reaction times failed to accomplish complete reduction in one step. If the product was purified by distillation without the second reduction, the yield of pure diimine was
9.1 g. (
71%). The submitters report that on ten times the scale the yield with one reduction was
81 g. (
64%).
10.
The product obtained is analytically pure, and the submitters claim it is suitable for polymerization. It is also shown to be homogeneous by GC.
3. Discussion
1,10-Diazacycloöctadecane has also been prepared by another general method which involves reaction of
1,8-dibromoöctane and
N,N'-ditosyl-1,8-octanediamine with
potassium carbonate as base, followed by cleavage of the sulfonamide groups;
2 the yield, however, is not as good as that by the present method. The present procedure is essentially a scale-up of the general method used by Stetter and Marx
3 for preparing a series of macrocyclic diimines of 10- to 21-membered rings, with a 25–78% yield for the crucial cyclization step. The procedure is adaptable for even larger scale preparations, using 5 moles of the acid chloride and a 100-gallon reaction kettle.
The reaction is generally applicable, to further extension, for making macrobicyclic diamines
4 with bridgehead
nitrogen atoms by using monocyclic diimines and an appropriate acid chloride. The main difference in the procedures is that
triethylamine is used as the
hydrogen chloride acceptor in the cyclization step instead of using an extra mole of diimine.
Triethylamine hydrochloride does not interfere with purification in this case since the bicyclic diamides are fairly soluble in
benzene but insoluble in water. The bicyclic diamides are isolated by first removing
triethylamine hydrochloride from the reaction mixture by filtration and water washings, followed by evaporation of the solvent. If
triethylamine is used as the base in the preparation of monocyclic dilactams, the products are invariably contaminated with
triethylamine hydrochloride, and the yields of the pure products are lower.
The same sequence of reactions are used to prepare macrobicyclic diamines with polyether linkages.
5,6 The reduction of macrobicyclic diamides can be accomplished with diborane using the procedure described by Brown and Heim,
7 but the present procedure is less involved and gives good yields.
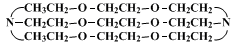
Appendix
Chemical Abstracts Nomenclature (Collective Index Number);
(Registry Number)
ethanol (64-17-5)
potassium carbonate (584-08-7)
hydrogen chloride (7647-01-0)
Benzene (71-43-2)
diethyl ether (60-29-7)
hydrogen (1333-74-0)
sodium hydroxide (1310-73-2)
silver nitrate (7761-88-8)
nitrogen (7727-37-9)
sodium (13966-32-0)
Triethylamine hydrochloride (554-68-7)
Tetrahydrofuran (109-99-9)
lithium aluminum hydride (16853-85-3)
N,N-dimethylformamide (68-12-2)
suberyl dichloride (10027-07-3)
triethylamine (121-44-8)
fluorocarbon (593-53-3)
octamethylenediamine dihydrochloride
1,10-Diazacyclooctadecane (296-30-0)
1,10-Diazacyclooctadecane-2,9-dione (56369-11-0)
N,N'-ditosyl-1,8-octanediamine
1,8-diaminooctane
1,8-dibromooctane
Copyright © 1921-, Organic Syntheses, Inc. All Rights Reserved