Org. Synth. 1978, 58, 113
DOI: 10.15227/orgsyn.058.0113
NUCLEOPHILIC α-sec-AMINOALKYLATION: 2-(DIPHENYLHYDROXYMETHYL)PYRROLIDINE
[2-Pyrrolidinemethanol, α,α-diphenyl-, (±)-]
Submitted by D. Enders, R. Pieter, B. Renger, and D. Seebach
1.
Checked by C. Hutchins and M. F. Semmelhack.
1. Procedure
Caution! Since N-nitrosopyrrolidine is a potent carcinogen and is produced as an intermediate, this entire "one-pot" procedure should be performed in a well-ventilated hood. Wearing of disposable polyethylene gloves is recommended.
Benzene has been identified as a carcinogen; OSHA has issued emergency standards on its use. All procedures involving benzene should be carried out in a well-ventilated hood, and glove protection is required.
A dry,
250-ml., one-necked, round-bottomed flask equipped with a
magnetic stirrer and a
three-way stopcock is charged with
4 g. (0.05 mole) of ethyl nitrite (Note 1),
4 g. of dry tetrahydrofuran (Note 2), and
2.35 g. (0.0331 mole) of pyrrolidine (Note 3). The stopcock is closed
(Note 4), and the mixture is stirred at room temperature for 2 days. Excess
ethyl nitrite,
tetrahydrofuran, and the
ethanol formed are removed from the
N-nitrosopyrrolidine (Note 5) by stirring at 25° under reduced pressure (
10 mm., water aspirator,
(Note 6) for 2 hours. The stopcock is fitted with a
rubber septum, the air in the system is replaced with dry
argon (Note 4) and
(Note 7)), and
50 ml. of tetrahydrofuran is injected by syringe. A solution of
lithium diisopropylamide is prepared in a separate, dry,
100-ml. flask by adding
21.1 ml. (0.0340 mole) of a 1.61 M solution of n-butyllithium in hexane (Note 8) to a solution of
3.44 g. (4.76 ml., 0.0341 mole) of diisopropylamine (Note 9) in
25 ml. of tetrahydrofuran at −78° (
methanol–dry ice bath) with stirring under
argon. The solution is warmed to 0° in 15 minutes, then added dropwise with a syringe within 4 minutes to the nitrosamine solution, stirred at −78°. Stirring of the yellow to orange solution is continued at this temperature for 25 minutes. A solution of
5.46 g. (0.0300 mole) of benzophenone in 12 ml. of tetrahydrofuran is added dropwise by syringe
(Note 10), and the mixture is stirred for 12 hours at −78°, then warmed to 0° within 2 hours. After addition of 0.6 ml. (0.03 mole) of water, the flask is transferred from the
argon line to a
rotary evaporator (within the hood). Solvents and
diisopropylamine are removed under reduced pressure in a 40° bath
(Note 11). The remaining solid is dissolved with slight warming in
120 ml. of dry methanol (Note 12), before
3.9 g. (66 equivalents) of Raney nickel (Note 13) is rinsed into the solution with
30 ml. of dry methanol. The reaction vessel is equipped again with the three-way stopcock, and the air in the flask is replaced with
hydrogen (Note 7). The flask is filled five times with
hydrogen from a balloon; during this operation vigorous stirring of the
Raney nickel–
methanol suspension is necessary. The flask is attached to a
mercury bubbler to maintain a positive
hydrogen pressure (200 mm.) supplied from a cylinder, as shown in
Figure 1. The reaction mixture is stirred for 3 hours at room temperature while a slow stream of
hydrogen is passed through the system. The major part of the solution is decanted and filtered, and the remaining
Raney-nickel suspension is extracted by refluxing three times for 10 minutes each with
20 ml. of methanol (Note 14). The combined
methanol solutions are concentrated under reduced pressure. The residue is dissolved in
150 ml. of diethyl ether and 100 ml. of water, the layers are separated
(Note 15), and the aqueous layer is extracted three times with
50-ml. portions of ether. The combined extracts are dried over
sodium carbonate and concentrated in a rotary evaporator to a total volume of 150 ml. Dry
hydrogen chloride gas is bubbled into the solution with stirring until the mixture is acidic, The almost colorless precipitate of the hydrochloride is filtered, washed two times with
30-ml. portions of dry ether, and dried in a
desiccator under reduced pressure for 3 hours, giving
5.99–6.11 g. (
58–60%, based on
benzophenone) of the product, m.p.
244–249° (dec.). Recrystallization from
methanol–acetone gives
5.06–5.20 g. (
58–60%) of analytically pure product, m.p.
267–269° (dec.)
(Note 16). The free base is obtained by treatment of the hydrochloride with
10% aqueous sodium hydroxide and extraction with
ether, m.p.
82–83° (Note 17).
Figure 1.
2. Notes
1.
Ethyl nitrite was prepared as described in Org. Synth., Coll. Vol. 2, 204 (1943), or purchased from Merck-Schuchardt and distilled before use, b.p.
17°. The volatile nitrite can be easily handled as a
50% tetrahydrofuran solution and stored in a refrigerator.
2.
Technical grade tetrahydrofuran, available from BASF-A G or Fisher Scientific Company, was dried by distillation, first from
potassium hydroxide then from
lithium aluminum hydride and used for all operations in this procedure. For a warning note regarding the purification of tetrahydrofuran, see
Org. Synth., Coll. Vol. 5, 976 (1973).
3.
Pyrrolidine, b.p. 87–88°, obtained from Aldrich Chemical Company, Inc., or BASF-A G was distilled from
potassium hydroxide before use.
5.
Nitrosamines are strong carcinogens
2,3;
N-nitrosopyrrolidine causes liver tumors in rats.
2,4 Although the one-pot procedure described here prevents contact with the nitrosamine, utmost care must be used to avoid contact with the reaction mixture during all manipulations.
6.
At the beginning of the evacuation the pressure should be lowered slowly to prevent bumping.
7.
This was done by alternately evacuating and filling with dry
argon three times; during the reaction a pressure of about 50 mm. above atmospheric was maintained using a mercury bubbler.
8.
Purchased from Metallgesellschaft, Frankfurt, or Alfa-Products, Division of the Ventron Corporation. The content of the solution was determined prior to use by acidimetric titration.
9.
The
diisopropylamine, b.p. 83–84°, available from Fluka A G, BASF-A G, or Aldrich Chemical Company, Inc., was purified by refluxing over
potassium hydroxide and subsequent distillation. It was stored over
calcium hydride.
10.
Benzophenone, m.p. 47–49°, was purchased from Riedel-de-Haen-A G or from Fisher Scientific Company. The reaction mixture turns green and then blue during the addition, because of the formation of ketyl radicals.
11.
The checkers found it more convenient to remove the volatile material at this stage by warming the stirred mixture at 40° and using a water aspirator vacuum (10 mm.). About 3 hours were required.
12.
Methanol was dried by heating at reflux for 3 hours over
magnesium, then distilling.
13.
The
Raney nickel reagent was prepared by addition of
9.5 g. of sodium hydroxide pellets over 8–10 minutes to a stirred suspension of 7.8 g of nickel–aluminum alloy (50% Ni, 50% Al powder, purchased from Merck-Schuchardt) in 120 ml. of distilled water, contained in a
250-ml. beaker. Fifteen minutes after the addition was completed, the beaker was immersed into a 70°
water bath for 20 minutes. The water was decanted, and the catalyst was washed sequentially with two 20-ml. portions of distilled water and two
20-ml. portions of methanol.
14.
Caution! The dry Raney nickel catalyst is pyrophoric. The residues can be destroyed by allowing them to ignite and burn on filter paper in a safe place.15.
Upon dissolving the residue in
200 ml. of ether and 100 ml. of water, the checkers obtained an emulsion that cleared slowly on standing for 2–3 hours.
16.
The literature reports m.p.
>240°,
5 >250°,
6 and
262–263°.
7 The yield given includes a small second crop obtained by recrystallization of the filtrate residue. The reported yield and m.p. data were obtained by the checkers. The submitters report
6.50–6.95 g. (
75–80%, m.p.
260–265°) before recrystallization and
5.20–5.62 g. (
60–65%, m.p.
267–269°) for analytically pure product.
17.
The m.p. is reported to be
81–82°6 and 83°.
5 The spectral properties are: IR spectrum (KI) cm
−1: strong absorptions at 3360, 3080, 3060, 3020, 2980–2800, 1595, 1490, 1450, 1400, 1190, 1100, 1060, 1030, 990, 900, 750, 700, 660, and 635;
1H NMR (CDCl
3), δ (multiplicity, number of protons): 1.60 (m, 4H), 2.95 (m, 2H, with overlapping broad peak for O
H and N
H), 4.18 (m, 1H), 7.00–7.65 (m, 10H). The compound has psychostimulating activity.
7
3. Discussion
The procedure described here is an example of the "nitrosamine method" for the electrophilic substitution of
1 to
3,
via the intermediate anion
2, as outlined in detail in a recent review article.
8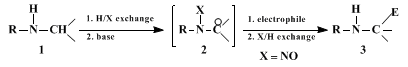
Currently, this is the only method that allows reversible enhancement of the acidity of α-nitrogen C-protons in a large variety of secondary amines, using nitroso-substituted nitrogen (X = NO, see examples in Table I). Nucleophile
2 is synthetically equivalent to α-aminocarbanion
4, while the inherent reactivity of a
carbon adjacent to an amino nitrogen is electrophilic (see the immonium ion
5).
8,9 Lithiated nitrosamines are also useful because their formation and reactions with electrophiles occur in high yield, as well as with a high degree of regio- and stereoselectivity (see Table I). For further information see the review article
8 and other recent publications.
10 This method has the disadvantage of carcinogenic nitrosamine intermediates, but this one-pot procedure reduces the danger of contact.
This preparation is referenced from:
TABLE I
α-SUBSTITUTED SECONDARY AMINES VIA ELECTROPHILIC SUBSTITUTION8
|
|
Starting Materials
|
Nitrosamine
|
Metallation Time (minutes)
|
Producta
|
Yield(%)
|
|
|
Benzaldehyde, dimethylamine
|
|
10
|
|
80
|
|
Benzylbromide, methylisopropylamine
|
|
10
|
|
80
|
|
Benzophenone, methylisopropylamine
|
|
10
|
|
75
|
|
Piperonal, methyl tert-butylamine
|
|
10
|
|
80
|
|
Carbon dioxide, methyl tert-butylamine
|
|
10
|
|
75b
|
|
Benzaldehyde, diethylamine
|
|
10
|
|
75b
|
|
Benzaldehyde, dihexylamine
|
|
10
|
|
80b
|
|
Benzophenone, azetidine
|
|
7
|
|
75b
|
|
Benzophenone, piperidine
|
|
180
|
|
50
|
|
Cyclohexanone, piperidine
|
|
180
|
|
55
|
|
Iodopropane, 3-hydroxypiperidine
|
|
240
|
|
40
|
|
Benzophenone, perhydroazepine
|
|
20
|
|
80b
|
|
a Isolated and characterized as hydrochlorides.
|
b Overall yield of stepwise procedure; the denitrosation was performed by bubbling gaseous hydrogen chloride into a benzene solution of the nitrosamine. This cleavage of nitrosamines is usually not as clean as the one with Raney nickel.
|
Appendix
Chemical Abstracts Nomenclature (Collective Index Number);
(Registry Number)
Raney-nickel
ethanol (64-17-5)
hydrogen chloride (7647-01-0)
Benzene (71-43-2)
methanol (67-56-1)
ether,
diethyl ether (60-29-7)
hydrogen (1333-74-0)
sodium hydroxide (1310-73-2)
magnesium (7439-95-4)
Cyclohexanone (108-94-1)
sodium carbonate (497-19-8)
carbon dioxide (124-38-9)
benzaldehyde (100-52-7)
nickel,
Raney nickel (7440-02-0)
acetone (67-64-1)
carbon (7782-42-5)
potassium hydroxide (1310-58-3)
Benzophenone (119-61-9)
piperidine (110-89-4)
ethyl nitrite (109-95-5)
diethylamine (109-89-7)
dimethylamine (124-40-3)
Benzylbromide (100-39-0)
n-butyllithium (109-72-8)
piperonal (120-57-0)
Tetrahydrofuran (109-99-9)
lithium aluminum hydride (16853-85-3)
pyrrolidine (123-75-1)
hexane (110-54-3)
nitrosamine (35576-91-1)
argon (7440-37-1)
calcium hydride (7789-78-8)
methylisopropylamine (4747-21-1)
Azetidine (503-29-7)
lithium diisopropylamide (4111-54-0)
diisopropylamine (108-18-9)
2-(Diphenylhydroxymethyl)pyrrolidine,
2-Pyrrolidinemethanol, α,α-diphenyl-, (±)- (112068-01-6)
N-nitrosopyrrolidine (930-55-2)
Iodopropane (107-08-4)
3-hydroxypiperidine (6859-99-0)
perhydroazepine (111-49-9)
methyl tert-butylamine (14610-37-8)
dihexylamine (143-16-8)
Copyright © 1921-, Organic Syntheses, Inc. All Rights Reserved