Replaced
Org. Synth. 1977, 56, 65
DOI: 10.15227/orgsyn.056.0065
METHYL (E)-3-NITROACRYLATE
[2-Propenoic acid, 3-nitro-, methyl ester (E)-]
Submitted by John E. McMurry
1, John H. Musser, Ian Fleming
2, J. Fortunak, and C. Nübling.
Checked by Wayland E. Noland and David D. McSherry.
1. Procedure
Caution! Part A should be carried out in an efficient fume hood to protect the operator from poisonous nitrogen dioxide vapors.
A. Methyl 2-iodo-3-nitropropionate. A dry, 5-l., three-necked, round-bottomed flask is fitted with a mineral oil-containing liquid-sealed mechanical stirrer, a gas-inlet tube connected to a drying tower containing 1:1 sodium hydroxide–Drierite, and an open neck that will later be connected to a rubber septum. The flask is flushed gently by passing dry nitrogen through the drying tower and gas-inlet tube as methyl acrylate (298.4 g., 331 ml., 3.47 moles) (Note 1) and anhydrous diethyl ether (2500 ml.) (Note 1) are added through the open neck. The flask is fitted with the rubber septum and then cooled to 0° by covering three-fourths of the flask with ice in a large ice bath made from a picnic cooler insulated on top with glass wool. The gas-inlet tube is removed just long enough to permit the addition of iodine (250 g., 0.98 mole) (Note 1). The mixture is stirred for about 15 minutes and then dinitrogen tetroxide (76.6 ml., 1.24 moles) (Note 1) and (Note 2) is introduced rapidly with a syringe (Note 3) through the rubber septum. The reaction solution is stirred at 0° for 30 hours. The solution is divided into three batches of about 1-l., which are cooled in a freezer to −4°. One batch at a time is removed and added to a 2-l. separatory funnel where it is washed with 80 ml. of aqueous 70% saturated sodium sulfite solution that had been precooled to −4° (Note 4). The ethereal solutions are washed alternately so that each batch is exposed to room temperature for the minimum time. The sodium sulfite washes are repeated two or three times on each batch until the color of the ethereal solution remains light yellow when kept at −4° (Note 5). The cooled ethereal solutions are then dried by washing each with precooled (to −4°) aqueous solutions containing 80 ml. of saturated sodium chloride and 15 ml. of saturated sodium sulfite. The ethereal solutions are then combined in a 4-l. Erlenmeyer flask and dried over anhydrous sodium sulfate at −4° overnight in a freezer. The dried solution is then filtered to remove the sodium sulfate, and concentrated by evaporating it in portions in a 1-l. round-bottomed flask on a rotating evaporator fitted with a cold-finger condenser containing a dry-ice bath (Note 6). The solution should be concentrated until it contains approximately equal volumes of product and ether. The solution is then cooled to −78° for 30 minutes (Note 6) and (Note 7), causing the separation of 403.5–409 g. of ether–wet crystals. The crystals are filtered and stored in a freezer, and the filtrate is eluted through a chromatographic column containing 700 g. of silica gel (Baker Analyzed, 60–200 mesh, 7-cm. column diameter), using 30% diethyl ether/petroleum ether (b.p. 30–60°) as eluent. The solution containing the first band to be eluted, which is yellow, is evaporated on a rotating evaporator fitted with a cold finger condenser cooled with dry ice as described above. This gives a residue of 35–54.5 g. of yellowish crystals, which are a 7:3 mixture of methyl 2-iodo-3-nitropropionate and methyl (E)-3-nitroacrylate. The ether-wet product from the original filtration and the eluted product are combined in a 1-l. round-bottomed flask and dried at 0° under a vacuum (0.3–0.5 mm.) for 3–4 hours, giving 396–401 g. (78–79%), m.p. 32–35°, of methyl 2-iodo-3-nitropropionate containing a small amount of methyl (E)-3-nitroacrylate. This mixture may be used directly in the next step. The product can be recrystallized from 30% ether/petroleum ether (b.p. 30–60°) to obtain thermally unstable white plates, m.p. 33–35° (Note 8).
B. Methyl (E)-3-nitroacrylate. Anhydrous diethyl ether (2800 ml.) is placed in a dry 5-l. three-necked, round-bottomed flask fitted with a mineral-oil, liquid-sealed, mechanical stirrer and a drying tower. The contents are cooled to 0° by covering three-fourths of the flask with ice in a large ice bath made from a picnic cooler insulated on top with glass wool. Powdered anhydrous sodium acetate (95 g., 1.16 moles) (Note 9) is added to the flask, followed by methyl 2-iodo-3-nitropropionate (150 g., 0.579 mole). The flask that contained the ester is rinsed with cold anhydrous diethyl ether (200 ml.), which is then added to the 5-l. flask. The flask is stoppered and its contents are stirred vigorously at 0° for 40 hours (Note 10). The flask is then removed from the ice bath and may be kept, until needed, in a freezer for 5–7 days. The ethereal solution is then decanted and filtered to remove the sodium acetate. The residue in the flask is rinsed with three 150-ml. portions of ether; the rinse solutions are also filtered and added to the original ethereal solution. The ethereal solution is then divided into three batches (about 1-l. each), each of which is added successively to a 2-l. separatory funnel. Each batch is washed with three 80-ml. portions of aqueous 70% saturated sodium hydrogen carbonate solution, followed by one wash with 80 ml. of saturated brine. The ether solutions are then combined in a 4-l. Erlenmeyer flask and dried overnight over anhydrous sodium sulfate in a freezer at −4°. The solution is filtered to remove the drying agent and then evaporated in portions in a 1-l. round-bottomed flask on a rotating evaporator fitted with a cold-finger condenser cooled with dry ice as described in Part A. The yellow, solid residue is dissolved in a minimum of 50% diethyl ether/petroleum ether (b.p. 30–60°) and cooled to −20° for 30 minutes (Note 7). The yellow crystals are collected by filtration and washed with cold diethyl ether. The filtrate and ether wash are concentrated and cooled to −70° for 30 minutes, giving a small second crop of crystals. The combined yield of methyl (E)-3-nitroacrylate is 67–69 g. (88–91% from dried methyl 2-iodo-3-nitropropionate) as yellow plates, m.p. 33–35° (Note 11).
Caution! Methyl (E)-3-nitroacrylate is a potent lachrymator, like nitroolefins in general. Contact with the eyes, or accidental transfer from hands to eyes, should be avoided. If lachrymation occurs, the eyes should be washed thoroughly with water until the irritation stops.
2. Notes
1.
The submitters used the following reagents as supplied:
iodine, ether, and sodium acetate from the Mallinckrodt Chemical Works;
dinitrogen tetroxide from Matheson Gas Products, Inc.; and
methyl acrylate from the Aldrich Chemical Company, Inc. The checkers obtained their reagents from the same sources, except that the
iodine (resublimed) was obtained from Aldrich. The
dinitrogen tetroxide was purchased in a lecture bottle.
2.
Dinitrogen tetroxide is often contaminated with
dinitrogen trioxide, giving the condensed liquid a blue-green color. This can be oxidized by passing dry air through the condensed liquid at −10° for 1–2 hours until the characteristic amber-brown color of
dinitrogen tetroxide persists.
3.
Slower introduction of
dinitrogen tetroxide by either dropwise addition of an ethereal solution or entrainment in a stream of
nitrogen gas gives similar results. The direct injection method was found to be easiest. The syringe should be precooled in a dry-ice bath to avoid back pressure during the
dinitrogen tetroxide additions.
4.
The
sodium sulfite solution is made by preparing a saturated solution at room temperature, cooling it to −4° and decanting it from the resulting precipitate, and diluting with cold water to 70% saturation.
5.
If kept at higher temperatures, the solution will darken to red and eventually brown due to thermal decomposition with release of
iodine.
6.
Diethyl ether was used as the liquid phase of the slurry in the dry-ice baths.
7.
The low crystallization temperatures require the use of a drying tower connected to the crystallization flask to prevent condensation of atmospheric moisture.
8.
1H NMR (60 mHz, CDCl
3): δ 3.81 (s, 3H, OC
H3), 4.56–5.25 (m, 3H, C
H2C
H).
9.
The anhydrous
sodium acetate (purchased from the Aldrich Chemical Company, Inc.) should be dried by heating at 135° overnight (12–16 hours), cooled in a
vacuum desiccator, and then ground to a fine powder which is again dried in a vacuum desiccator for several hours.
10.
The progress of the reaction can be monitored by
1H NMR analysis of concentrated aliquots obtained by evaporation of the solvent and following the appearance of the
vinyl signals of the product at δ 6.95 and 7.55
(Note 12) and the disappearance of the A
2B multiplet of the starting material at δ 4.56–5.25
(Note 8).
11.
The progress of the reaction can be monitored by
1H NMR analysis of concentrated aliquots obtained by evaporation of the solvent and following the appearance of the vinyl signals of the product at δ 6.95 and 7.55
(Note 12) and the disappearance of the A
2B multiplet of the starting material at δ 4.56–5.25
(Note 8).
12.
1H NMR (60 MHz, CDCl
3): δ 3.8 (s, 3H, OC
H3), 6.95 (d,
J = 14 hz, 1H, =C
HNO
2-), 7.55 (d,
J = 14 Hz, 1H, =C
HNO
2): IR (CCl
4): cm.
−1 3100 s, 3000 m, 2950 s, 2880 m (all CH), 1720 s (C=O), 1640 ms (C=C), 1530 s and 1350 s (NO
2).
3. Discussion
This procedure for the synthesis of
methyl (E)-3-nitroacrylate replaces the procedure for the corresponding ethyl ester given in Volume 56 of this series.
3 Both procedures were adapted from those for
methyl (E)-3-nitroacrylate of Stevens and Emmons
4 for the addition and of Schechter, Conrad, Daulton, and Kaplan
5 for the elimination. Four major changes have been made in the procedures: (1) rapid introduction of
dinitrogen tetroxide, (2) limited purification of the
methyl 2-iodo-3-nitropropionate, (3) use of finely powdered anhydrous
sodium acetate, and (4) carrying out the elimination at 0° for 40 hours followed by 5–7 days in a freezer. With these modifications, the preparation is reproducible and proceeds in 69–72% overall yield from
iodine.
The compound has also been prepared by the reaction of
methyl acrylate with
dinitrogen tetroxide and
oxygen in
ether at 0°, followed by hydrolysis (of the 2-nitrito and 2-nitrato esters), neutralization, and distillation, in 13% overall conversion.
6 The
methyl 2-hydroxy-3-nitropropionate obtained as a coproduct in 27% conversion was dehydrated by refluxing with
acetyl chloride to give additional
methyl (E)-3-nitroacrylate in 43% conversion.
6 The compound has also been prepared by the reaction of
methyl acrylate with
nitrosyl chloride in
ether in a
sealed tube at room temperature to give
methyl 2-chloro-3-nitropropionate in 32% yield, which was dehydrochlorinated with anhydrous
sodium acetate in
ether at −5° and +20° to
methyl (E)-3-nitroacrylate in 37% yield.
7The homolog,
ethyl (E)-3-nitroacrylate, has been shown to be an extremely reactive receptor in the Michael reaction. It has been used in the synthesis of the
α-methylenebutyrolactone moiety
8 characteristic of many sesquiterpenes, as shown below.
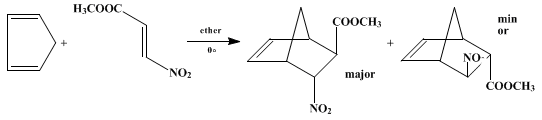
Methyl (E)-3-nitroacrylate is a reactive dienophile in the Diels-Alder reaction. With
cyclopentadiene it gives the corresponding adduct,
methyl 6-nitro-2-norbornene-5-carboxylate, in 43% yield in refluxing
benzene9 or in quantitive yield in
ether at 0°,
10 as shown below. The adduct was shown by NMR analysis to consist of an 86 : 14 mixture of
endo- :
exo-nitro stereoisomers, which could not be separated by crystallization.
10
Appendix
Chemical Abstracts Nomenclature (Collective Index Number);
(Registry Number)
petroleum ether
dinitrogen tetroxide
brine
2-Propenoic acid, 3-nitro-, methyl ester (E)-
freezer
dinitrogen trioxide
Benzene (71-43-2)
ether,
diethyl ether (60-29-7)
sodium acetate (127-09-3)
sodium sulfite (7757-83-7)
acetyl chloride (75-36-5)
sodium hydrogen carbonate (144-55-8)
sodium chloride (7647-14-5)
sodium sulfate (7757-82-6)
oxygen (7782-44-7)
nitrogen (7727-37-9)
iodine (7553-56-2)
nitrogen dioxide,
exo-nitro (10102-44-0)
nitrosyl chloride (2696-92-6)
methyl acrylate (96-33-3)
vinyl (2669-89-8)
CYCLOPENTADIENE (542-92-7)
Methyl 2-iodo-3-nitropropionate
methyl 2-hydroxy-3-nitropropionate
methyl 2-chloro-3-nitropropionate
α-methylenebutyrolactone (547-65-9)
methyl 6-nitro-2-norbornene-5-carboxylate
METHYL (E)-3-NITROACRYLATE (52745-92-3)
ethyl (E)-3-nitroacrylate
Copyright © 1921-, Organic Syntheses, Inc. All Rights Reserved