Org. Synth. 1986, 64, 19
DOI: 10.15227/orgsyn.064.0019
α-AMINO ACETALS: 2,2-DIETHOXY-2-(4-PYRIDYL)ETHYLAMINE
[4-Pyridineethanamine, β,β-diethoxy]
Submitted by John L. LaMattina and R. T. Suleske
1.
Checked by Paul Hebeisen and Andrew S. Kende.
1. Procedure
It has been reported by workers at Pfizer Global R&D that oxime tosylates in this series are thermally very sensitive at temperatures slightly above their melting points (Chem. Eng. News 2001, October 1). It has also been reported that the 3-isomer demonstrated low-level shock sensitivity (Tetrahedron Lett. 1999, 40. 6739).
While this sensitivity was not observed during the checking of this procedure, extreme caution should be taken when handling these materials.
A. 4-Acetylpyridine oxime. Hydroxylamine hydrochloride (25.0 g, 0.36 mol) (Note 1) is dissolved in 50 mL of water, and the solution is added to 70 mL of 20% aqueous sodium hydroxide in a 500-mL Erlenmeyer flask. To this magnetically stirred solution is added at one time 4-acetylpyridine (36.3 g, 0.30 mol) (Note 2); a precipitate forms rapidly. The reaction mixture is stirred at 0–5°C for 2 hr; then the precipitate is collected by suction filtration and washed with 500 mL of cold water.
The product, (mp 122–146°C, 33–36 g, 81–88%) can be shown from its 1H NMR spectrum (Note 3) to be a 5:1 mixture of the E- and Z-isomers of 4-acetylpyridine oxime. To obtain pure E-isomer (Note 4), the product is recrystallized twice as follows. The crude product is dissolved in 600 mL of hot water in a 2-L Erlenmeyer flask, the hot solution is decanted from any undissolved residue, and the supernatant liquid is allowed to cool slowly to 30°C during 2–3 hr by placing the flask on a cork ring. The precipitate is collected at this temperature by suction filtration. A second crystallization by the same procedure yields pure E-oxime, which is dried under reduced pressure over Drierite to constant weight. The yield of E-4-acetylpyridine oxime, mp 154–157°C (Note 5), is 27.1–28.3 g (66–69%).
B. 4-Acetylpyridine oxime tosylate. Pure E-oxime (27.1 g, 0.20 mol) and p-toluenesulfonyl chloride (47.9 g, 0.22 mol) (Note 6) are added to 100 mL of anhydrous pyridine (Note 7) in a 1-L, round-bottomed flask fitted with a drying tube and a large magnetic stirring bar. The reaction mixture is stirred at 25°C for 24 hr; a precipitate of pyridine hydrochloride forms. A 500-mL portion of ice water is added with continued stirring. the initial precipitate dissolves, and a voluminous white precipitate soon forms. This is collected by suction filtration, washed with three 150-mL portions of cold water and dried under reduced pressure and over Drierite to constant weight. The yield of pure tosylate, mp 79–81°C (Note 8), is 55.1 g (95%).
C. 2,2-Diethoxy-2-(4-pyridyl)ethylamine. To a 2-L, round-bottomed flask containing 80 mL of absolute ethanol (Note 9) and fitted with a magnetic stirrer and a reflux condenser with a drying tube, potassium metal (7.60 g, 0.19 mol) is slowly added (Note 10). When the metal has dissolved, the solution is cooled to 0–5°C and (E)-4-acetylpyridine tosylate (55.1 g, 0.19 mol) dissolved (with gentle warming) in 320 mL of absolute ethanol is added over 15 min through a dropping funnel to the stirred solution at 0–5°C. During this period a precipitate of potassium p-toluenesulfonate forms. The temperature of the stirred mixture is allowed to rise to room temperature for 1 hr. The mixture is diluted with 1 L of anhydrous ether and filtered by suction. The precipitate is quickly washed with 150 mL of anhydrous ether. The ether filtrates are combined, and hydrogen chloride gas is bubbled through the ether solution for 15 min. A precipitate forms immediately. The precipitate is collected by suction filtration, washed with three 170-mL portions of anhydrous ether, and dried briefly under reduced pressure. The dihydrochloride thus obtained is dissolved in 200 mL of water, and powdered sodium carbonate is added until the mixture reaches a pH of >10. The mixture is extracted four times with 125-mL portions of chloroform. The combined chloroform extracts are dried over anhydrous magnesium sulfate and concentrated at reduced pressure to an oil. This orange–red oil is distilled at 0.2 mm to yield 29.7 g (74.5%) of the amine as a colorless oil, bp 93–95°C (Note 11).
2. Notes
1.
Hydroxylamine hydrochloride 97% (mp 155–157°C), available from Aldrich Chemical Company, Inc. or Fisher Scientific Company, is suitable for use without further purification.
2.
4-Acetylpyridine (98%) from Aldrich Chemical Company, Inc. was distilled under reduced pressure (bp
103–104°C/14–16 mm) prior to use.
3.
In
dimethyl sulfoxide-d6, the
E and
Z isomers show OH proton resonances at δ 11.65 and 10.97, respectively.
4.
Use of the isomer mixture prevents isolation of oxime tosylate in crystalline form at the next step and leads to reduced overall yield of pure amine.
5.
The lit.
2 melting point for the oxime is
158°C.
6.
p-Toluenesulfonyl chloride was purified prior to use by the procedure of L. Fieser and M. Fieser in "Reagents for Organic Synthesis."
37.
Pyridine AR (Mallinckrodt, Inc.) was used directly.
8.
The lit.
2 mp for this compound is
80°C.
9.
Ethanol was dried by reflux over
magnesium ribbon.
11.
This compound has the following 90-MHz
1H NMR spectrum (CDCl
3) δ: 0.75 (br s, 2 H, NH
2), 1.20 [t, 6 H,
J = 7, (CH
3CH
2O)
2], 2.97 (s, 2 H, CH
2NH
2), 3.41 [m, 4 H, (OCH
2CH
3)
2], 7.37 (d of d, 2 H,
J = 2, 4.5, pyridine H
3 and H
5), 8.58 (d of d, 2 H,
J = 2, 4.5, pyridine H
2 and H
6). Anal. calcd. for C
11H
18N
2O
2: C, 62.83; H, 8.63; N, 13.32. Found: C, 62.63; H, 8.52; N, 13.20.
3. Discussion
α-Amino ketones are useful intermediates for the preparation of a variety of heterocycles, including imidazoles,
4 oxazoles,
5 and pyrazines.
6 Unfortunately, pyrazine formation can be a complicating side reaction because of the tendency of α-amino ketones to dimerize. One way to avoid this problem is to generate these intermediates in a protected form, specifically, as α-amino acetals.
7 Such derivatives allow one to manipulate the amino moiety as desired. The acetal can then be hydrolyzed at the appropriate interval to complete the synthesis.
α-Amino acetals can be prepared from α,α-dialkoxynitriles either via catalytic hydrogenation
8 or by reaction with organometallic reagents.
9 However, these methods are limited by the availability of the appropriate starting material. The procedure here offers a more simple approach that involves the Neber rearrangement. Although this reaction is generally used to prepare α-amino ketones, use of an anhydrous
ethanol medium readily results in acetal formation. A summary of other α-amino acetals prepared using this procedure appears in Table I.
This reaction, like all Neber rearrangements, is limited by the availability of the appropriate oxime tosylate.
10 Substrates in which the aryl group contains an electrondonating function are unstable, since they have a propensity to undergo Beckmann rearrangement. However, this difficulty can be resolved by subsequent conversion of the α-amino acetals. For example, catalytic hydrogenation of
2,2-diethoxy-2-(p-bromophenyl)ethylamine yields the known parent compound
2,2-diethoxy-2-phenylethylamine; (these two α-amino acetals readily undergo hydrolysis and should be protected from moisture). Other approaches to α-aminoacetals include reduction of α-azidoacetals,
11 reaction of α-haloketimines with alcohol,
12 and reaction of
N,N-dichloroamines with
sodium ethoxide.
13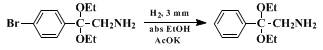
TABLE I
PREPARATION OF α-AMINO ACETALS
|
|
|
|
Ar
|
R
|
Yield (%)
|
bp (°C/mm)
|
mp (°C), HCl salt
|
|
|
2-Pyridyl
|
H
|
58
|
82/0.2
|
150 (dec)a
|
|
3-Pyridyl
|
H
|
53
|
84/0.2
|
187–188a
|
|
4-Pyridyl
|
CH3b
|
40
|
98/0.2
|
129–130a
|
|
4-O2N-C6H4
|
H
|
78
|
—c
|
116 (dec)
|
|
4-Br-C6H4
|
H
|
92
|
—d
|
|
|
|
|
bFor the preparation of this material in Step C, gaseous HCl is bubbled into the ethereal filtrate for 3 hr. Presumably the longer reaction time is necessary for steric reasons.
|
cThis material decomposes on distillation and is purified by column chromatography (silica gel–chloroform).
|
dThis material decomposes on distillation and hydrolyzes when chromatographed on silica gel. However, 1H-NMR analysis indicates that it is >95% pure.
|
Appendix
Chemical Abstracts Nomenclature (Collective Index Number);
(Registry Number)
Drierite
dimethyl sulfoxide-d6
ethanol (64-17-5)
hydrogen chloride,
HCl (7647-01-0)
ether (60-29-7)
sodium hydroxide (1310-73-2)
chloroform (67-66-3)
magnesium (7439-95-4)
sodium carbonate (497-19-8)
pyridine (110-86-1)
sodium ethoxide (141-52-6)
potassium (7440-09-7)
Hydroxylamine hydrochloride (5470-11-1)
magnesium sulfate (7487-88-9)
pyridine hydrochloride (628-13-7)
p-Toluenesulfonyl chloride (98-59-9)
2,2-Diethoxy-2-(4-pyridyl)ethylamine,
4-Pyridineethanamine, β,β-diethoxy (74209-44-2)
4-acetylpyridine (1122-54-9)
4-acetylpyridine oxime (1194-99-6)
2,2-diethoxy-2-(p-bromophenyl)ethylamine
2,2-diethoxy-2-phenylethylamine
potassium p-toluenesulfonate
E-4-acetylpyridine oxime
4-Acetylpyridine oxime tosylate (74209-52-2)
Copyright © 1921-, Organic Syntheses, Inc. All Rights Reserved