Org. Synth. 1990, 69, 44
DOI: 10.15227/orgsyn.069.0044
erythro-DIRECTED REDUCTION OF A β-KETO AMIDE: ERYTHRO-1-(3-HYDROXY-2-METHYL-3-PHENYLPROPANOYL)PIPERIDINE
[Piperidine, 1-(3-hydroxy-2-methyl-1-oxo-3-phenylpropyl)-, (R*, R*)-(±)-]
Submitted by M. Fujita and T. Hiyama
1.
Checked by Gregory P. Roth and Albert I. Meyers.
1. Procedure
A.
1-(2-Benzoylpropanoyl)piperidine. A
dry, 300-mL, two-necked, round-bottomed flask is equipped with a
magnetic stirrer and charged with
nitrogen. One neck is connected to a
three-way stopcock equipped with a balloon filled with
nitrogen, and the other neck is capped with a
rubber septum. The flask is charged with
100 mL of anhydrous tetrahydrofuran (THE) (Note 1) and
10.1 g (14.1 mL, 0.100 mol) of diisopropylamine (Note 2) and immersed in an
acetone–dry ice bath. A
1.68 M hexane solution of butyllithium (60 mL, 0.10 mol) (Note 3) is added dropwise with stirring over a 10-min period, and the stirring is continued for 1 hr at −78°C. To the resulting
lithium diisopropylamide (LDA) solution is added dropwise
14.1 g (0.100 mol) of propanoylpiperidine (Note 4) with stirring over a 10-min period, and the stirring is continued for 2 hr at −78°C
(Note 15). The rubber septum is replaced with a
polyvinyl chloride (or Teflon) tube connected to another 300-mL, two-necked, round-bottomed flask, which is equipped with a magnetic stirrer and a three-way stopcock, charged with
100 mL of anhydrous THF and
13.5 g (16.3 mL, 0.110 mol) of benzoyl chloride (Note 6), and immersed in an acetone–dry ice bath. The balloon is taken off and
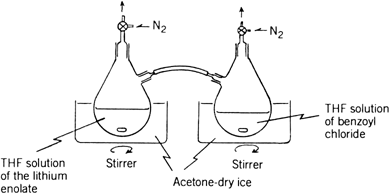
nitrogen is passed through the two stopcocks so that the reaction mixture does not come in contact with air (see the apparatus shown in
Fig. 1). By inclining the first flask, the THF solution of the
lithium enolate of 1-propanoylpiperidine is added to the
THF solution of benzoyl chloride in the second flask through the polyvinyl chloride tube over a 5-min period. After the solution is stirred for 0.5 hr at −78°C, it is allowed to warm to room temperature, diluted with
200 mL of dichloromethane, and washed with 200 mL of water. The organic layer is separated, and the aqueous layer is extracted with two
50-mL portions of diethyl ether. The combined organic layers are dried over anhydrous
magnesium sulfate and concentrated with a
rotary evaporator. Recrystallization from
diethyl ether–hexane affords
12.5 g (
51%) of
1-(2-benzoylpropanoyl)piperidine, mp
100–101°C (Note 7).
Figure 1
B. erythrol-1-(3-Hydroxy-2-methyl-3-phenylpropanoyl)piperidine. A 300-mL, two-necked, round-bottomed flask is equipped with a magnetic stirrer and charged with nitrogen. One neck is connected with a balloon charged with nitrogen, and the other neck is capped with a rubber septum. Into the flask are placed 50 mL of trifluoroacetic acid (Note 8) and 11.9 g of 1-(2-benzoylpropanoyl)piperidine (48.7 mmol) prepared as described in Part A; then the flask is immersed in an ice–water bath. To the flask is added 7.3 g of dimethylphenylsilane (8.24 mL, 54 mmol) (Note 9) over a 5-min period with the aid of a 10-mL syringe, and the resulting mixture is stirred for 4 hr in the ice bath. The mixture is diluted with 200 mL of dichloromethane and washed with 200 mL of water. After the organic layer is separated, the aqueous layer is extracted with two 50-mL portions of diethyl ether, and the combined organic layers are concentrated with a rotary evaporator (Note 10). The crude oil is placed in a 200-mL, one-necked flask and dissolved in 100 mL of methanolic 1 M sodium hydroxide. The solution is stirred for 1.5 hr at ambient temperature with a magnetic stirrer. The mixture is diluted with 200 mL of dichloromethane and washed with 50 mL of water. The organic layer is separated, and the aqueous layer is extracted with two 50-mL portions of diethyl ether. The combined organic layers are dried over anhydrous magnesium sulfate and concentrated by rotary evaporation (Note 11). The residual oil is subjected to column chromatography using 100 g of silica gel (Note 12). After the first fraction (800t mL) of hexane is eluted, the second fraction, eluted with 500 mL of diethyl ether, is collected and concentrated. Recrystallization of the resulting oil from diethyl ether–hexane gives 10.2 g of material, mp 85–86°C. The yield is 90% (Note 13).
The analogous threo derivatives can be made by use of tris(diethylamino)sulfonium difluorotrimethylsilicate as the catalyst (Note 14).
2. Notes
1.
Tetrahydrofuran (THF) is freshly distilled over
benzophenone ketyl.
2.
Diisopropylamine is distilled over
calcium hydride.
3.
The
hexane solution of butyllithium is purchased from Wako Pure Chemicals Industries, Ltd. and titrated before use.
4.
Propanoylpiperidine is prepared from
propanoyl chloride and
piperidine according to a similar procedure described in
2.
5.
The
lithium enolate of (2-benzoylpropanoyl)piperidine should be handled below −20°C, as it decomposes above 0°C.
6.
Benzoyl chloride of commercial grade is distilled before use.
7.
Spectral characteristics are as follows:
1H NMR (CDCl
3) δ: 1.46 (d, 3 H,
J = 7.2), 1.3–1.7 (m, 6 H), 3.25–3.65 (m, 4 H), 4.40 (q, 1 H,
J = 7.2), 7.25–7.65 (m, 3 H), 7.85–8.05 (m, 2 H); IR (KBr) cm
−1: 1696, 1620, 1450, 1204, 686; MS (50 eV)
m/z rel intensity, 245 (M
+; 14), 140 (37), 105 (100), 84 (99), 77 (47). Anal. calcd. for C
15H
19NO
2: C, 73.44; H, 7.81; N, 5.71. Found: C, 73.22; H, 7.87; N, 5.69.
8.
Trifluoroacetic acid was purchased from Aldrich Chemical Company, Inc. (also available from Tokyo Kasei Co. LTD, Japan), and used directly.
9.
Dimethylphenylsilane was purchased from Aldrich Chemical Company, Inc. (also available from Shin-etsu Kagaku Co. LTD, Japan), and used directly.
10.
About half of the product is trifluoroacetylated during the concentration procedure.
11.
A 400-MHz
1H NMR analysis of the crude oil showed exclusive formation of the erythro isomer of the material (>99:1).
12.
A
glass column (35 mm × 20 cm) packed with Wakogel C-200 is used.
13.
Spectral characteristics are as follows:
1H NMR (CDCl
3) δ: 1.03 (d, 3 H,
J = 7), 1.3–1.7 (m, 6 H), 2.84 (dq, 1 H,
J = 2.5, 3), 3.2–3.7 (m, 4 H), 4.30 (broad s, 1 H), 5.06 (d, 1 H,
J = 2.5), 7.2–7.4 (m, 5 H); IR (KBr) cm
−1: 3350, 1606; MS (relative intensity)
m/z, 247 (M
+; 7), 232 (20), 141 (100), 112 (26), 84 (43), 79 (20). Anal. calcd. for C
15H
21NO
2: C, 72.84; H, 8.56; N, 5.66. Found: C, 72.74; H, 8.69; N, 5.52.
14.
threo-1-(3-Hydroxy-2-methyl-3-phenylpropanoyl)piperidine. A 300-mL, two-necked, round-bottomed flask is equipped with a magnetic stirrer and charged with dry
nitrogen. One neck is connected with a three-way stopcock, one arm of which is connected to a balloon filled with
nitrogen. The other neck is capped with a rubber septum. The flask is evacuated with a vacuum pump under heating with a heat gun and
nitrogen is admitted. This operation is repeated 3 times to replace the inner atmosphere of the flask completely with dry
nitrogen. In the flask are placed
50 mL of hexamethylphosphoric triamide (Note 15),
12.3 g of 1-(2-benzoylpropanoyl)piperidine (50 mmol), and
8.2 g of dimethylphenylsilane (9.2 mL, 60 mmol) by syringe, and then the flask is immersed in an ice–water bath. To the flask is added dropwise
2.5 mL of a 1 M tetrahydrofuran (THF) solution of tris(diethylamino)sulfonium difluorotrimethylsilicate (TASF) (2.5 mmol) (Note 16) with the aid of a syringe, and the resulting mixture is stirred for 6 hr at ice-bath temperature. In order to complete the reaction,
3.4 g of dimethylphenylsilane (3.8 mL, 25 mmol) and
1.5 mL of a 1 M THF solution of TASF (1.5 mmol) are added and stirring is continued for an additional 6 hr at the same temperature. The mixture is quenched with
50 mL of 1 M hydrochloric acid, stirred for 1.5 hr at ambient temperature, and extracted with three
100-mL portions of diethyl ether. The organic layer is washed with 50 mL of water, dried over anhydrous
magnesium sulfate, and concentrated with a rotary evaporator. The crude oil is subjected to column chromatography using
100 g of silica gel (Note 17). After the first fraction, eluted with
800 mL of hexane, is removed, the second fraction, eluted with
500 mL of diethyl ether, is concentrated (Note 18). Recrystallization of the residue from
diethyl ether–
hexane gives
8.03 g (
65%) of material, mp
79–80°C (Note 19). The mother liquor is concentrated and again subjected to column chromatography
(Note 20) to give the same material, which, after recrystallization from
diethyl ether–
hexane, melts at
77–79°C (
1.5 g,
12%). The total yield amounts to
77%.
15.
Hexamethylphosphoric triamide is distilled from
calcium hydride under reduced pressure of
nitrogen. In place of
hexamethylphosphoric triamide,
1,3-dimethyl-3,4,5,6-tetrahydro-2(1H)pyrimidinone (DMPU), which is dried and purified similarly,
3 can be used.
16.
TASF was prepared according to the procedure described in
4. Typically,
diethylamino(trimethyl)silane (6.4 g, 8.3 mL, 44 mmol) is added drop by drop under a dry inert atmosphere to an ethereal solution
(20 mL) of diethylaminosulfur trifluoride (DAST, purchased from Aldrich Chemical Company, Inc., and used directly) (3.2 g, 2.4 mL, 20 mmol) under cooling with a dry ice–acetone bath. The mixture is allowed to warm to room temperature and stirred for 72 hr at room temperature. The initial homogeneous solution separates into two layers. The upper layer is removed with the aid of a syringe. The lower layer is washed with dry
ether (10 mL × 3) and dried under reduced pressure to afford TASF as a solid (
6.0 g, 16.6 mmol,
83% yield). All the isolation operations should be carried out under an inert atmosphere such as
nitrogen. The solid is dissolved in
THF to give a 1
M solution (the volume of the solution is 16.6 mL) which is stored under a dry
nitrogen atmosphere.
17.
A glass column (35 mm × 20 cm) packed with Wakogel C-200 is used.
18.
A 400-MHz
1H NMR analysis of the crude oil showed exclusive formation of the threo isomer of the material (>99%).
19.
Spectral characteristics are as follows:
1H NMR (CDCl
3) δ: 1.22 (d,
J = 7.3 H), 1.1–1.7 (m, 6 H), 2.8–3.8 (m, 5 H), 4.7–4.8 (m, 2 H), 7.31 (s, 5 H); IR (KBr) cm
−1: 3380, 1606; MS (relative intensity)
m/z 247 (M
+; 6), 232 (16), 141 (100), 112 (23), 84 (39), 79 (15). Anal. calcd. for C
15H
21NO
2: C, 72.84; H, 8.56; N, 5.66. Found: C, 72.70; H, 8.63; N, 5.65.
20.
A glass column (20 mm × 25 cm) packed with 50 g of Wakogel C-200 is used. After the first fraction, eluted with
300 mL of dichloromethane, was removed, the second fraction, eluted with
300 mL of dichloromethane–diethyl ether (1 : 4), was concentrated.
3. Discussion
Aldols of the
erythro configuration are prepared by aldol condensation of various metal enolates.
5 6 7 An alternative approach is reduction of β-keto esters
8 or amides
9 with
zinc borohydride. The hydrosilane-based reduction described here provides
erythro aldols under high stereocontrol and is practical because of the mild conditions and easy handling of readily available hydrosilanes.
3 The scope of this reduction is summarized in Table I. No epimerization at the chiral center is observed as shown in the last entry. The erythro-selective reduction with the PhMe
2SiH/CF
3COOH reagent is also applicable to the reduction of 2-oxy or 2-amino ketones.
10,11
TABLE I
erythro-SELECTIVE REDUCTION OF α-SUBSTITUTED β-KETO ACID DERIVATIVES WITH PhMe2SiH/H+ REAGENTa
|
|
Substrateb
|
Time, hr
|
Productc
|
% Yieldd
|
Threo: Erythroe
|
|
|
|
3
|
|
94
|
2:98
|
|
|
20
|
|
89
|
1:99
|
|
|
3
|
|
99
|
1:99
|
|
|
3
|
|
87
|
1:>99
|
|
|
4
|
|
98
|
1:>99
|
|
aCarried out on a 0.5–1.0-mmol scale at 0°C employing PhMe2SiH (1.2 mol equiv) and CF3COOH (1–2 mL/mmol).
|
bRacemates were employed unless noted.
|
cMajor isomers are shown.
|
dPurified by silica gel chromatography.
|
eThe ratio was determined by 90 or 400 MHz 1H NMR analysis.
|
fThe optically pure substrate was prepared according to a known method: see Evans, D. A.; Ennis, M. D.; Le, T.; Mandel, N.; Mandel, G. J. Am. Chem. Soc. 1984, 106, 1154.
|
Preparation of
threo aldols is sometimes a problem. For stereoselective synthesis by aldol condensation, propionate esters of mesitol must be employed.
12 A general, alternative approach to
threo aldols is
threo-directed reduction of β-keto esters.
13 Although the stereoselectivity of this reduction is usually low, reduction of β-keto amides with
potassium triethylborohydride (KBHEt3) is extremely selective.
14 The hydrosilane/F
− reduction of β-keto amides provides
threo aldols of high diastereomeric purity when aroyl-substituted amides are employed.
3 The scope of this reduction is summarized in Table II. High
threo selectivity is observed only for reduction of 2-aroylpropanoates, whereas the reduction of 2-alkanoylpropanoates proceeds with poor selectivity and gives
erythro isomers as the major product.
15 The hydrosilane/F
− reduction is also applicable to the
threo-selective reduction of α-oxy and α-amino ketones.
10
TABLE II
THREO-SELECTIVE REDUCTION OF β-KETO AMIDES WITH PhMe2SiH/F− REAGENTa
|
|
Substrateb
|
Time hr
|
Productc
|
% Yieldd
|
Threo: Erythroe
|
|
|
|
12
|
|
98
|
>99:1
|
|
|
16
|
|
86
|
99:1
|
|
|
16
|
|
92
|
99:1
|
|
|
22
|
|
93
|
23:27
|
|
aCarried out on a 0.5–1.0-mmol scale at 0°C employing PhMe2SiH (1.2 mol equiv) and TASF (10 mol %).
|
bRacemates were employed.
|
cMajor isomers are shown.
|
dPurified by silica gel chromatography.
|
eThe ratio was determined by 90 or 400 MHz 1H NMR analysis.
|
Appendix
Chemical Abstracts Nomenclature (Collective Index Number);
(Registry Number)
silica gel
benzophenone ketyl
erythro-1-(3-Hydroxy-2-methyl-3-phenylpropanoyl)piperidine
Piperidine, 1-(3-hydroxy-2-methyl-1-oxo-3-phenylpropyl)-, (R*, R*)-(±)-
lithium enolate of 1-propanoylpiperidine
THF solution of benzoyl chloride
erythrol-1-(3-Hydroxy-2-methyl-3-phenylpropanoyl)piperidine
lithium enolate of (2-benzoylpropanoyl)piperidine
threo-1-(3-Hydroxy-2-methyl-3-phenylpropanoyl)piperidine
TASF
hydrochloric acid (7647-01-0)
ether,
diethyl ether (60-29-7)
sodium hydroxide (1310-73-2)
nitrogen (7727-37-9)
benzoyl chloride (98-88-4)
piperidine (110-89-4)
dichloromethane (75-09-2)
magnesium sulfate (7487-88-9)
propanoyl chloride (79-03-8)
butyllithium (109-72-8)
Tetrahydrofuran,
THF (109-99-9)
hexane,
hexane solution of butyllithium (110-54-3)
calcium hydride (7789-78-8)
trifluoroacetic acid (76-05-1)
hexamethylphosphoric triamide (680-31-9)
lithium diisopropylamide (4111-54-0)
diisopropylamine (108-18-9)
Diethylaminosulfur trifluoride (38078-09-0)
diethylamino(trimethyl)silane (996-50-9)
1,3-dimethyl-3,4,5,6-tetrahydro-2(1H)pyrimidinone (7226-23-5)
propanoylpiperidine
1-(2-Benzoylpropanoyl)piperidine (99114-34-8)
dimethylphenylsilane (766-77-8)
tris(diethylamino)sulfonium difluorotrimethylsilicate (59201-86-4)
zinc borohydride
potassium triethylborohydride
Copyright © 1921-, Organic Syntheses, Inc. All Rights Reserved