Org. Synth. 1990, 68, 162
DOI: 10.15227/orgsyn.068.0162
3-METHYL-2(5H)-FURANONE
[2(5H)-Furanone, 3-methyl-]
Submitted by Jan H. Näsman
1
Checked by Alan T. Johnson and James D. White.
1. Procedure
Caution! Hydrogen peroxide attacks the skin and may decompose violently. The first step should be carried out behind a safety screen, and the operator should wear safety glasses and rubber gloves. Air must not be admitted to the hot distillation residue in Step 2.
A. 2(5H)-Furanone. A 6-L, three-necked, round-bottomed flask equipped with two condensers, a dropping funnel, and a 12 × 55-mm magnetic stirring bar is charged with 480 g (5 mol) of furfural (Note 1) and 2.0 L of methylene chloride. The addition of 200 g of sodium sulfate (Note 2) and 150 g of N,N-dimethylethanolamine (Note 3) in one portion each is followed immediately by 460 g of formic acid (Note 4), carefully added in portions over a period of 2 min, after which 100 mL of 30% hydrogen peroxide (Note 5) is added in one portion. The mixture is stirred vigorously. After 5 min the mixture will reflux and another 800 mL of 30% hydrogen peroxide is added dropwise during 9 hr (Note 6) while stirring is continued. When the addition is complete, the mixture is vigorously stirred as long as it refluxes and then stirred gently overnight. The organic phase is separated, and the water phase is extracted with the 200 mL of methylene chloride that is used to wash out residues from the reaction flask.
The methylene chloride phase is washed with two 150-mL portions of saturated sodium disulfite solution (Note 7) and dried over magnesium sulfate and sodium sulfate. After a negative peroxide test (Note 8), the solvent is removed. The crude product (255 g) is fractionated through a 30-cm Vigreux column. The material boiling at 85–85°C (13 mm) is collected to give 210 g of butenolide, which is yellow because of some high-boiling residues. Redistillation through the 30-cm Vigreux column and collection of the material boiling at 100–102°C (30 mm), 95–97°C (19 mm), 89–91°C (16 mm), or 79–81°C (9 mm) gives colorless butenolide. In this way 170.2 g (41%) of pure butenolide is obtained.
B. Furyl phosphorodichloridate. A 1-L flask, protected from moisture by a calcium chloride tube, is charged with 42 g (0.5 mol) of 2(5H)-furanone, 85 g (0.55 mol) of phosphoryl chloride, and 100 mL of methylene chloride. A solution of 65 g (0.5 mol) of ethyldiisopropylamine in 60 mL of methylene chloride is added dropwise during 4 hr at ambient temperature (Note 9). The resulting mixture is stirred overnight (12 hr), after which 6.5 g of the amine in 10 mL of methylene chloride is added in one portion and stirring is continued for 20 hr (Note 10). The solvent is removed on a rotary evaporator and 200 mL of dry ether (Note 11) is added cautiously, followed by 100 mL of pentane (in that order), to the dark residue to precipitate the amine hydrochloride. The flask is stoppered and shaken for 1–2 min. The hydrochloride is filtered by suction and washed immediately with 100 mL of dry ether and 200 mL of pentane or petroleum ether (Note 12). The bottle is tightly stoppered and the filtrate is allowed to stand in the refrigerator (+4°C) overnight. The clear brown ethereal phase is decanted from a dark lower phase, and the solvent is evaporated. The residue (ca. 100 g) is distilled at the water pump. In order to obtain pure, color-stable, yellow dichloridate it is usually necessary to distill it twice. The first distillation is done rapidly, collecting the material that boils at 73–98°C (9 mm) to give 65–75 g of product, which usually darkens within a few days (Note 13) and (Note 14). Redistillation (Note 15), collecting the material that boils at 91–93°C (22 mm), 88–90°C (16 mm), or 73–76°C (9 mm), gives 60–65 g of pure product (Note 16). The yield is 60–65%.
C. Furyl N,N,N',N'-tetramethyldiamidophosphate. To 180 mL of dry diethyl ether, chilled to −30°C, is added 56.7 g (4.2 equiv) (1.26 mmol) of dimethylamine (Note 17). This solution is added—during 1–2 hr from a double-jacketed dropping funnel, protected from moisture by a calcium chloride tube and connected to a cryostat regulated to −30°C—to a stirred mixture (Note 18) of 60 g (0.30 mol) of the freshly distilled furyl phosphorodichloridate and 250 mL of ether in a two-necked, 1-L flask equipped with a condenser, protected from moisture by a calcium chloride tube and connected to the cryostat. This flask is chilled in an ice bath during the addition of the first 2 equiv of the dimethylamine. After the addition of dimethylamine is complete, stirring is continued for 20 hr while the mixture is warmed on a water bath at 35°C. The hydrochloride that forms is carefully filtered off with suction and washed with two 70-mL portions of dry ether. The combined ether phases are evaporated to give ca. 65 g (99%) of crude product. Distillation, discarding a yellow forerun and collecting the fraction boiling at 149–152°C (20 mm) or 131–134°C (7 mm) (Note 19), affords 52–58 g (79–88%) of pure material (Note 20).
D. 3-Methyl-2(5H)-furanone. To 10.9 g (50 mmol) of furyl tetramethyldiamidophosphate in 90 mL of tetrahydrofuran (THF) (Note 21), chilled to −75°C, is added 21.9 mL (55 mmol) of a 2.51 M hexane solution of butyllithium (Note 22) at a rate (6–10 min) such that the temperature reaches −60°C but does not exceed this level. The resulting mixture is chilled to −75°C for 10 min; then 8.9 g (63 mmol) of methyl iodide in 20 mL of tetrahydrofuran is added with a syringe during 7–8 min (Note 23) so that the temperature does not rise above −55°C. After the addition is complete, the temperature is raised to 0°C and the mixture is concentrated to ca. 40 mL. Water (30 mL) and ethyl acetate (50 mL) are added, the phases are separated, and the dark inorganic phase is extracted with two 50-mL portions of ethyl acetate. The combined, yellow organic phases are washed with brine and dried over magnesium sulfate. The solvent is evaporated to give 10.5 g of crude 2-(3-methylfuryl) tetramethyldiamidophosphate, which need not be purified for the next reaction (Note 24).
To the phosphate in a 250-mL flask on a water bath at 25°C (Note 25) is added 20 mL of 98–100% formic acid (Note 26), and the resulting mixture is stirred until bubbling has ceased (30–40 min). Benzene (50 mL) is added and most of the excess formic acid is removed on an evaporator. To the residue are added 50 mL of ethyl acetate and 30 mL of a sodium chloride–sodium carbonate solution (Note 27). The organic phase is washed twice with the latter solution (i.e., a total of 3 × 20 mL), the combined inorganic phases are extracted once with 50 mL of ethyl acetate, the combined organic phases are dried over magnesium sulfate, the solvent is removed, and the product is distilled to give 3.2 g (64%) of 3-methyl-2(5H)-furanone, bp 97–101°C (19 mm).
2. Notes
1.
Practical-grade furfural from Fluka Chemical Corporation or Aldrich Chemical Company, Inc. was used without any purification. Very dark
furfural can be used, but it foams at the beginning of the reaction and leads to lower yields.
2.
Sodium sulfate is used to salt out the water phase;
brine is not effective. The yield without the sulfate is 5–10% lower.
3.
N,N-Dimethylethanolamine (99% pure) was obtained from EGA CHEMIE or Aldrich Chemical Company, Inc. The role of the compound is to isomerize any
2(3H)-furanone formed.
4.
Formic acid (98–100%), obtained from Merck & Company, Inc., was used.
5.
"Perhydrol" (30%), obtained from Merck & Company, Inc., gave reproducible results without efforts to determine the activity of the
peroxide. An excess is used.
6.
The process is a fine balance between oxidation and isomerization of the initially formed
2(3H)-furanone. Longer addition times produce better yields; however, the benefit is of marginal value.
7.
Sodium disulfite, Na2S2O5, from Merck & Company, Inc. was used. The saturated solution of disulfite should be the lower phase.
8.
The mixture is tested for
peroxide as follows. Prepare an approximately
1% solution of ferrous ammonium sulfate. Transfer 5 mL to each of two test tubes and add
0.5 mL of 0.5 M sulfuric acid and
0.5 mL of 0.1 M potassium thiocyanate solution to each tube. Add
5 mL of the methylene chloride solution to one of the test tubes and shake well. The aqueous phase in the
methylene chloride tube should not develop a brown-red color when examined parallel to the blank.
9.
Phosphoryl chloride from Fluka Chemical Corporation or Aldrich Chemical Company, Inc., and
methylene chloride (purum) from Merck & Company, Inc., were used. Unless the contents of a freshly opened bottle were used,
methylene chloride was distilled from
phosphorus pentoxide (20 g/L) before use. The
amine (Fluka or Aldrich) was distilled from and stored over
potassium hydroxide. The best yields were obtained with once-recovered
amine.
10.
The reaction is reversible. In order to obtain pure (
97–98%) dichloridate it is essential to add the 6.5 g of
amine after the first equivalent has reacted.
11.
Ordinary
diethyl ether is stored over
calcium chloride for 36 hr, filtered, and dried over sodium wire.
12.
The use of more
pentane or
petroleum ether gave a product of better stability and purity.
13.
Once-distilled product was usually not color-stable for prolonged periods.
14.
The distillation flask is allowed to cool before air is passed into it. A vigorous polymerization may occur if air is passed into the hot residue, which may be safely discarded after the addition of
acetone (an exothermic, but easily controlled reaction). Spectroscopic data for
furyl phosphorodichloridate are as follows:
1H NMR (60 MHz, CDCl
3, TMS) δ: 5.85 (m, 1 H, furan-
H3), 6.30 (m, 1 H, furan-H4), 7.05 (m, 1 H, furan-H5);
13C NMR (CDCl
3, TMS) δ: 92.5 (
3JPC = 7, furan-C3), 111.5 (
4JPC = 3, furan-C4), 137.1 (
4JPC = 3, furan-C5), 147.9 (
2JPC = 12, furan-C2). MS
m /
e [relative intensity (rel. int.)]: 202 (16), 200 (26), 119 (4), 117 (6), 83 (100), 55 (31). M
+ 201.9160: calcd. 201.9167 for C
4H
3Cl
2O
3P; observed 199.9195. calcd. 199.9197. IR cm
−1: 1610 (s), 1300 (s), 980 (s), 890, 870. Anal. calcd. for C
4H
3Cl
2O
3P: C, 23.9, H, 1.5. Found: C, 23.8, H, 1.5.
15.
Pure, pale-yellow dichloridate is stable for months without extensive change of color if stored in
well-stoppered bottles in the refrigerator.
16.
The purity of this product is
97–98%. It contains some
butenolide; therefore an excess of
dimethylamine is used in the subsequent step.
17.
Dry
dimethylamine from Fluka Chemical Corporation or MC and B Manufacturing Chemists was used as delivered.
18.
A
12 × 55-mm heavy magnetic stirring bar is used for good stirring.
19.
The monochloroamidate distills at 123°C (9 mm) and is identified in the
1H NMR by its
3JPH = 13.5.
Distill slowly in the beginning! The purity of the product is ≥99%. Redistill if a dark-yellow color develops; however, this color does not precluded successful lithiation.
20.
The distilled diamide is a pale-yellow oil at room temperature; it freezes in the refrigerator (+4°C) if seeded within some hours. The first spontaneous crystalization took several weeks. It can also be obtained as snow-white crystals from
diisopropyl ether/hexane, mp
15–16°C. Spectroscopic data for
furyl tetramethyldiamidophosphate are as follows:
1H NMR (400 MHz, CDCl
3 TMS) δ: 2.71 (d, 12 H,
3JPH = 10, two N(CH
3)
2), 5.62 (m, 1 H, furan-H3), 6.28 (m, 1 H, furan-H4), 6.95 (m, 1 H, furan-H5);
13C NMR (15.03 MHz, CDCl
3, TMS) δ: 151.9 (d,
2JPC = furan-C2), 134.5 (s, furan-C5), 111.3 (s, furan-C4), 88.8 (d,
3JPC = 4, furan-C3), 36.6 (d,
2JPC = 4, N(CH
3)
2). Note that multiplicities s and d refer to C-P coupling. MS
m/
e (rel. int.): 218 (6), 136 (6), 135 (100), 127 (2), 111 (2), 92 (7), 90 (2), 83 (4), 69 (3). M
+ at 218.0822: calcd. 218.0820 for C
8H
12N
2O
3P; IR cm
−1: 2900, 2800, 1610, 1300, 990, 960.
21.
Tetrahydrofuran (Merck) was distilled from
sodium–
benzophenone ketyl prior to use.
22.
Butyllithium was obtained from Aldrich Chemical Company, Inc.; and
methyl iodide, from Merck & Company, Inc. Butyllithium was titrated with
phenanthroline as indicator prior to use according to the method of Watson and Eastham.
2 Fresh alkoxide-free
butyllithium should be used to ensure pure product.
23.
Methyl iodide should be added carefully in the beginning when the reaction mixture is mostly solid.
24.
The phosphate can be crystallized from
diisopropyl ether/
hexane at −20°C in
80–85% yield; mp
42–44°C. Spectroscopic data for
2-(3-methylfuryl) tetramethyldiamidophosphate are as follows:
1H NMR (400 MHz, CDCl
3, TMS) δ: 1.95 (dxt, 3 H,
J = 0.4 and 2.2 CH
3), 2.73 (d, 12 H,
3JPH = 10.2, two N(CH
3)
2), 6.16 (dxdxq, 1 H,
J = 0.4 and 2.2, furan-H4), 6.91 (dxdxq, 1 H,
J = 0.4 and 2.2, furan-H5);
13C NMR δ: 8.4 (sxq, CH
3), 36.6 (dsq,
2JPC = 4, N(CH
3)
2), 98.7 (dxs,
3JPC = 5, furan-C3), 113.8 (dxs,
4JPC = 2, furan-C4), 133.9 (dxd,
4JPC = 2 furan-C5), 147.8 (dxs,
3JPC = 8, furan-C2); multiplicities underlined in the
13C spectrum refer to C-P coupling, the other to C-H coupling; MS
m/
e (rel. int.): 232 (7), 135 (100), 97 (3), 92 (5). M
+ at 232.0980: calcd. 232.0977 for C
9H
17N
2O
3P: Calcd. for C
9H
17N
2O
3P: C, 46.55, H, 7.33, N, 12.07. Found: C, 46.5, H, 7.6, N, 12.0.
25.
The water bath can be removed after 5 min. The reaction is vigorous in the beginning, and chilling is necessary to avoid formation of
dimethylformamide (DMF), which is formed at elevated temperatures.
26.
Formic acid (98–100%), obtain from Merck & Company, Inc., was used.
27.
Warning: CO2 evolution. The
sodium chloride–
sodium carbonate solution was prepared from
185 g of sodium chloride and
110 g sodium carbonate dissolved in water to give a total volume of 1 L.
3. Discussion
The preparation of
2(5H)-furanone is the scaled-up and slightly modified procedure
3 based on the report of Badovskaya that
furfural is oxidized with performic acid to give a mixture of furanones.
4 The preparation here is improved by the use of
N,N-dimethylaminoethanol as a catalyst for the isomerization of
2(3H)-furanone to
2(5H)-furanone. The complex between this amino alcohol and
formic acid does not enter the organic phase during workup, and the product is thus easily isolated simply by evaporation of the solvent.
The hitherto preferred method for preparation of the
butenolide is that of Price and Judge,
5 which can be modified (by extraction of the
bromolactone with
methylene chloride and elimination of
hydrogen bromide with
triethylamine or preferably with
diisopropylethylamine in
toluene at 70°C) of give routinely 60% or greater overall yield on a 6-mol scale. However, the large amount of
hydrogen bromide evolved is sometimes a nuisance, especially to inexperienced workers. The method reported here is fast and independent of scale (0.1–6 mol tried), the starting materials are cheap, and the product is easily isolated.
Substituted furfurals do not react at a synthetically useful rate when formic acid/ hydrogen peroxide is used. This suggests that the reaction takes place in the water phase and that substituted furfurals enter this phase only with difficulty.
The preparation of
furyl phosphorodichloridate is based on a method for preparation of
2-chlorofuran (16% yield, Hormi, Näsman, unpublished). Later the preparation was extended to a general method to prepare furyl esters from carboxylic acid chlorides lacking α-hydrogens and alkyl furyl carbonates from primary (other than methyl) and secondary alkyl chloroformates.
6 Phosphoryl chloride was the only acid chloride except carbon analogs found to give a furyl ester by the amine-catalyzed reaction.
Regioselective β-metallation for π-excessive five-ring heterocycles is not a novel reaction.
7 Oxazoline8 9 10 11 and
pyridine12 as well as carboxylate-
13 and carboxamide
14-substituted heterocycles have been lithiated. From the point of synthetic utility thiophenes have been shown to be useful substrates after careful optimization of reaction conditions; furans have been of less utility.
The generation of
2-(3-lithio)furyl tetramethyldiamidophosphate (≥95%) in
tetrahydrofuran with a slight excess of
butyllithium is a reliable procedure. The reagent usually forms a precipitate (active) when stored for prolonged times (3–12 hr) at −80°C and less than
35 mL of tetrahydrofuran/10 mmol of reagent is used.
7 The reagent darkens when warmed to −30°C. The reagent has been used on 100-mmol scales with no difficulties in methylation with
methyl iodide.
Methyl iodide is a very good electrophile for the reagent, whereas
ethyl iodide does not react. Other good substrates for this unmodified reagent are ketones, aldehydes, and chloromethyl ethers. Alkylation is difficult unless strongly activated substrates are use. For example,
benzyl chloride is unreactive,
benzyl bromide reacts but not completely, and the corresponding iodide gives a complex mixture.
The reagent must be added to the electrophile when the leaving group is an alkoxide. For example, quenching with MeOD on larger scales yields products labelled also in the 5-position, whereas reverse addition with good stirring does not.
The
furan products can be purified by flash chromatography
15 and should be used at once. A mixture of
ethyl acetate and
methylene chloride is a good solvent system for flash chromatography. Small residues of silica tend to partly decompose these furans within 2 weeks. The products are hygroscopic.
Diisopropyl ether–diethyl ether–hexane is a useful solvent system for recrystallization of solid furans.
The formic acid reaction to convert furans to butenolides seems to be general, although heating may be necessary for acceptor-substituted furans; dimethylformamide (DMF) is then a byproduct.
In conclusion,
furyl N,N,N',N'-tetramethylamidophosphate is the precursor to the
d3-synthon
1 for
butenolide, which is difficult to generate by direct or other indirect methods;
16 however,
17 for a metal–halogen exchange reaction of 3- or 4-bromo-2-methoxy- or 2-trimethylsiloxyfurans.
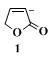
Appendix
Chemical Abstracts Nomenclature (Collective Index Number);
(Registry Number)
petroleum ether
amine
benzophenone ketyl
brine
dimethylformamide (DMF)
butenolide
2-(3-methylfuryl) tetramethyldiamidophosphate
Perhydrol
bromolactone
2-(3-Lithio)furyl tetramethyldiamidophosphate
calcium chloride (10043-52-4)
sulfuric acid (7664-93-9)
hydrochloride (7647-01-0)
Benzene (71-43-2)
ethyl acetate (141-78-6)
ether,
diethyl ether (60-29-7)
sodium chloride (7647-14-5)
hydrogen bromide (10035-10-6)
sodium carbonate (497-19-8)
sodium sulfate (7757-82-6)
formic acid (64-18-6)
acetone (67-64-1)
pyridine (110-86-1)
potassium hydroxide (1310-58-3)
toluene (108-88-3)
sodium (13966-32-0)
benzyl chloride (100-44-7)
hydrogen peroxide,
peroxide (7722-84-1)
Methyl iodide (74-88-4)
potassium thiocyanate (333-20-0)
Furan (110-00-9)
Furfural (98-01-1)
dimethylamine (124-40-3)
Pentane (109-66-0)
methylene chloride (75-09-2)
magnesium sulfate (7487-88-9)
Ethyl iodide (75-03-6)
benzyl bromide (100-39-0)
butyllithium (109-72-8)
Tetrahydrofuran (109-99-9)
diisopropyl ether (108-20-3)
ferrous ammonium sulfate (10045-89-3)
dimethylformamide (68-12-2)
hexane (110-54-3)
oxazoline
triethylamine (121-44-8)
N,N-dimethylethanolamine,
N,N-dimethylaminoethanol (108-01-0)
phenanthroline
2(5H)-Furanone (497-23-4)
phosphoryl chloride (10025-87-3)
diisopropylethylamine,
ethyldiisopropylamine (7087-68-5)
phosphorus pentoxide (1314-56-3)
sodium disulfite
Furyl phosphorodichloridate (105262-70-2)
furyl tetramethyldiamidophosphate,
Furyl N,N,N',N'-tetramethyldiamidophosphate,
furyl N,N,N',N'-tetramethylamidophosphate (105262-58-6)
2-chlorofuran (3187-94-8)
2(3H)-Furanone
3-Methyl-2(5H)-furanone,
2(5H)-Furanone, 3-methyl- (22122-36-7)
Copyright © 1921-, Organic Syntheses, Inc. All Rights Reserved