Org. Synth. 1992, 70, 10
DOI: 10.15227/orgsyn.070.0010
N-tert-BUTOXYCARBONYL-L-SERINE β-LACTONE AND (S)-3-AMINO-2-OXETANONE p-TOLUENESULFONIC ACID SALT
[(Carbamic acid, (2-oxo-3-oxetanyl)- 1,1-dimethylethyl ester, (S)-) and (2-Oxetanone, 3-amino-, (S)-, 4-methylbenzenesulfonate)]
Submitted by Sunil V. Pansare, Lee D. Arnold, and John C. Vederas
1.
Checked by P. S. Manchand, P. Mastrodonato-DeLora, and David L. Coffen.
1. Procedure
A.
N-(tert-Butoxycarbonyl)-L-serine β-lactone.
2 3 A
2-L, three-necked, round-bottomed flask is equipped with a
magnetic stirring bar, an
argon inlet adaptor, a low temperature
thermometer, and a
rubber septum (Note 1). The flask is charged with
tetrahydrofuran (1.1 L) and
triphenylphosphine (42.1 g, 160 mmol) (Note 2). The
triphenylphosphine is dissolved with stirring, and the flask is cooled to −78°C with a
dry ice-acetone bath maintained at that temperature
(Note 3). Distilled
diethyl azodicarboxylate (DEAD) (27.86 g, 160 mmol) is then added dropwise with a syringe over 10 min
(Note 4). The resulting pale yellow solution is stirred at −75°C to −78°C for 10 min, at which point a milky slurry is obtained. The rubber septum on the flask is quickly replaced with a
1-L, pressure-equalizing dropping funnel containing a solution of
N-(tert-butoxycarbonyl)-L-serine (32.8 g, 160 mmol) in
tetrahydrofuran (240 mL),
(Note 2) and
(Note 5) which is then added dropwise to the mixture over 30 min. After completion of the addition, the mixture is stirred at −75°C to −78°C for 20 min, the cooling bath is removed, and the mixture is slowly warmed with stirring to room temperature over 2.5 hr
(Note 6). The solvent is removed on a
rotary evaporator at 35°C. The residual pale yellow syrup is suspended in
hexane/
ethyl acetate (85/15, 20 mL), slurried, and the solid is removed by filtration. The filtrate is diluted with
ethyl acetate (30 mL) to give a solution which is applied to a 10 × 23-cm column of flash silica gel
4 (800 g) packed in
hexane/
ethyl acetate (85/15). The flask and the sides of the column are rinsed with additional
ethyl acetate (20 mL), which is added to the column that is then eluted with
hexane/
ethyl acetate (85/15, 2.7 L). The solvent is changed to
hexane/
ethyl acetate (7/3) and 500-mL fractions are collected. Concentration of fractions 6–12 on a rotary evaporator gives
12.07 (
40%) of pure
N-(tert-butoxycarbonyl)-L-serine β-lactone (Note 7) and
(Note 8).
B.
(S)-3-Amino-2-oxetanone p-toluenesulfonic acid salt.
5 A
500-mL, single-necked, round-bottomed flask is equipped with a magnetic stirring bar and an argon inlet adaptor
(Note 1). The flask is charged with a mixture of
N-(tert-butoxycarbonyl)-L-serine β-lactone (14.0 g, 74.8 mmol) and anhydrous
p-toluenesulfonic acid (13.5 g, 78.5 mmol) (Note 9). The argon inlet adaptor is replaced with a rubber septum and an argon inlet and the flask is cooled in an
ice bath for ca. 15 min. Anhydrous
trifluoroacetic acid (200 mL, (Note 10)) is added by
cannula along the sides of the flask over 20 min (stirring is initiated when possible). The pale yellow solution is stirred at 0°C for 10 min, the
trifluoroacetic acid is removed on a rotary evaporator at below 30°C, and the resulting syrup is placed under high vacuum (~0.2 mm) for ca. 1 hr. Anhydrous
ether (200 mL, (Note 11)) is added to the resulting solid, the mixture is triturated to break up lumps, and the suspension is filtered. The solid thus obtained is washed with
ether (100 mL), air dried (5 min), and then dried under reduced pressure (0.2 mm) overnight to give
18.4 g (
95% yield) of
(S)-3-amino-2-oxetanone p-toluenesulfonic acid salt (Note 12).
2. Notes
1.
The glass components of the apparatus are dried overnight in a 120°C
oven, and then assembled and maintained under an atmosphere of dry
nitrogen or
argon before use.
2.
Triphenylphosphine (obtained from Aldrich Chemical Company, Inc.) and
N-(tert-butoxycarbonyl)-L-serine (obtained from U.S. Biochemical Corporation) were dried over P
2O
5 for 72 hr and 24 hr, respectively.
Tetrahydrofuran was distilled from
sodium benzophenone ketyl directly into the glassware (under
argon) the day before and stored under
argon overnight until used.
3.
The temperature of the solution should be about −75°C before the
diethyl azodicarboxylate is added.
4.
Diethyl azodicarboxylate was obtained from Fluka AG. (
See warning,
p. 837). The material used was distilled, bp
82–83°C at 2 mm, in the
hood behind a safety shield. It is important that the addition of this compound to the reaction mixture be done at a constant rate without interruption because of its tendency to freeze in the syringe needle.
Dimethyl azodicarboxylate (17.7 mL, 160 mmol; δ = 1.33 g/mL at 25°C; bp
71–72°C at 2 mm) can be used in place of
diethyl azodicarboxylate. This compound is manfactured by Tokyo Kasei Kogyo Co. and is available from CTC Organics. The use of a very slight excess of
azodicarboxylate ester (a few percent relative to
triphenylphosphine) prevents reaction of
triphenylphosphine with the β-lactone product.
65.
The solution of dried
N-(tert-butoxycarbonyl)-L-serine was made up separately in an addition funnel under an atmosphere of
argon. This avoids complications that may arise if the funnel is prefitted on the reaction vessel.
6.
The reaction vessel was placed in a
water bath at room temperature after the temperature of the mixture was ca. 15°C.
7.
The reaction usually works better on a smaller scale (25 mmol) where a yield of 70% or greater can be obtained. The flash column was eluted such that the solvent level dropped 1 cm/13 sec. This corresponds to an approximate rate of 362 mL/min. The concentration and purification of the reaction mixture should be done as quickly as possible on the same day. Although storage of the concentrated reaction mixture at −20°C overnight results in substantial decomposition of the lactone, column fractions containing pure lactone (after chromatography) can be stored at 4°C overnight and concentrated on the following day.
The β-lactone is readily visualized by TLC (Merck, Kieselgel 60 F254, 0.25 mm thickness, hexane/ethyl acetate (65/35) as solvent system) by staining in iodine or by using bromocresol green spray (0.04% in EtOH, made blue by NaOH) followed by heating the plate to detect the lactone as a yellow spot on a blue background.
8.
The product exhibits the following properties: mp
119.5–120.5°C; [α]
24D −26.2° (CH
3CN,
c 1); IR cm
−1: 3358, 1836, 1678, 1533, 1290, 1104;
1H NMR (360 MHz, CD
2Cl
2) δ: 1.45 (s, 9 H, -C(CH
3)
3), 4.4–4.45 (m, 2 H, CH
2), 4.95–5.05 (m, 1 H, NH-CH), 5.2–5.4 (br s, 1 H, NH); MS (CI, NH
3), m/z 205 (M · NH
4⊕, 100%). Anal. Calcd for C
8H
13NO
4: C, 51.33; H, 6.99; N, 7.48. Found: C, 51.28; H, 7.01; N, 7.42.
9.
Anhydrous
p-toluenesulfonic acid was prepared from the monohydrate (obtained from Aldrich Chemical Company, Inc.) by solution in hot
benzene with the aid of
ethyl acetate and azeotropic distillation to 50% volume to remove water. The solution was then concentrated to a syrup on a rotary evaporator. The syrup was dissolved in a minimum volume of
acetone and excess
benzene was added to precipitate the anhydrous acid. This material was recrystallized from
acetone/
benzene and dried at 50°C for 12 hr. It was stored under reduced pressure over anhydrous CaSO
4. The material used melted at
104–105°C.
10.
Trifluoroacetic acid (obtained from Aldrich Chemical Company, Inc.) was refluxed over P
2O
5 for ca. 3 hr and then distilled from P
2O
5 under an atmosphere of
argon. The material used distilled at
68–71°C. All manipulations involving
trifluoroacetic acid (except removal on a rotary evaporator) were done in a fume hood.
11.
Commercially available anhydrous
ether (obtained from Fisher Scientific Company) was used directly from a freshly opened can.
12.
The product exhibits the following properties: mp (~4°C/min) 133–135°C (darkens), 173°C (dec) (rapid); [α]
24D −15.8° (DMF,
c 2.2); IR (Fluorolube mull) cm
−1: 3040, 1833, 1547;
1H NMR (360 MHz, d
7 DMF) δ: 2.3 (s, 3 H, ArCH
3), 4.66 (m, 1 H, CHHO), 4.74 (m, 1 H, CHHO), 5.54 (dd, 1 H, J = 4.6, 6.5, CH), 7.15 (d, 2 H, J = 8, m-ArH), 7.64–7.7 (d, 2 H, J = 8, o-ArH); FAB MS (glycerol) m/z 260 (MH
⊕). Anal. Calcd for C
10H
13NO
5S: C, 46.32; H, 5.05; N, 5.40; S, 12.37. Found: C, 46.44; H, 5.14; N, 5.24; S, 12.41.
Handling and Disposal of Hazardous Chemicals
The procedures in this article are intended for use only by persons with prior training in experimental organic chemistry. All hazardous materials should be handled using the standard procedures for work with chemicals described in references such as "Prudent Practices in the Laboratory" (The National Academies Press, Washington, D.C., 2011 www.nap.edu). All chemical waste should be disposed of in accordance with local regulations. For general guidelines for the management of chemical waste, see Chapter 8 of Prudent Practices.
These procedures must be conducted at one's own risk. Organic Syntheses, Inc., its Editors, and its Board of Directors do not warrant or guarantee the safety of individuals using these procedures and hereby disclaim any liability for any injuries or damages claimed to have resulted from or related in any way to the procedures herein.
3. Discussion
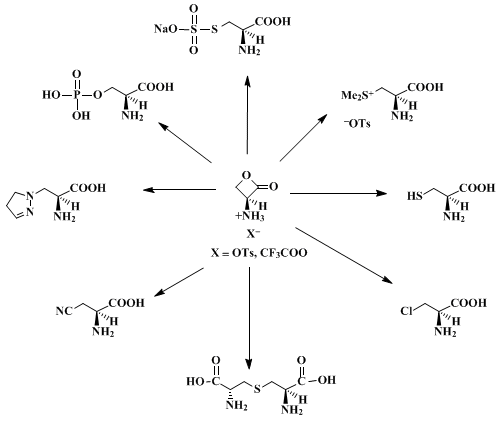
Recent work has shown that a large variety of carbon, nitrogen, oxygen, sulfur, and halogen nucleophiles attack chiral N-protected serine β-lactones at the β-carbon to give optically pure N-protected α-amino acids.
2,3,5 However, in certain cases (e.g., β-azidoalanine
7) these products are unstable to most common deprotection conditions. The procedure given here describes the preparation of
(S)-3-amino-2-oxetanone p-toluenesulfonic acid salt, a compound which reacts with a variety of nucleophiles to afford unprotected, optically pure α-amino acids directly (Scheme 1).
8 This salt has a long shelf life (many months) at room temperature provided that it is stored dry. It reacts rapidly with water (t
½ ~ 2.5 hr in unbuffered water; t
½ ~ 10 min in 50 mM
potassium phosphate at pH 6.8). However, good nucleophiles such as thiols afford high yields of sulfur-containing amino acids in water if the pH is kept at 5.0–5.5.
7 Since both enantiomers of
serine are relatively inexpensive, and
L-serine is readily available in isotopically labeled form, this approach should prove useful for syntheses of sensitive
D-amino acids as well as for preparation of the labeled
L-isomers.
Appendix
Chemical Abstracts Nomenclature (Collective Index Number);
(Registry Number)
benzophenone ketyl
2-Oxetanone, 3-amino-, (S)-, 4-methylbenzenesulfonate
Benzene (71-43-2)
ethyl acetate (141-78-6)
ether (60-29-7)
nitrogen (7727-37-9)
iodine (7553-56-2)
acetone (67-64-1)
sodium (13966-32-0)
D-amino (15194-15-7)
Tetrahydrofuran (109-99-9)
diethyl azodicarboxylate (1972-28-7)
hexane (110-54-3)
serine,
L-serine (56-45-1)
argon (7440-37-1)
trifluoroacetic acid (76-05-1)
triphenylphosphine (603-35-0)
p-toluenesulfonic acid (104-15-4)
dimethyl azodicarboxylate (2446-84-6)
(S)-3-AMINO-2-OXETANONE p-TOLUENESULFONIC ACID SALT (112839-95-9)
N-tert-Butoxycarbonyl-L-serine β-lactone,
N-(tert-Butoxycarbonyl)-L-serine β-lactone,
Carbamic acid, (2-oxo-3-oxetanyl)- 1,1-dimethylethyl ester, (S)- (98541-64-1)
N-(tert-Butoxycarbonyl)-L-serine
azodicarboxylate
potassium phosphate (7778-53-2)
(benzyloxycarbonyl)-β-(pyrazol-1-yl)-L-alanine
Copyright © 1921-, Organic Syntheses, Inc. All Rights Reserved