Org. Synth. 1996, 73, 152
DOI: 10.15227/orgsyn.073.0152
BIS(TRIFLUOROETHYL) (CARBOETHOXYMETHYL)PHOSPHONATE
[Acetic acid, [bis(2,2,2-trifluoroethoxy)phosphinyl]-, ethyl ester]
Submitted by Carl Patois
1, Philippe Savignac
1, Elie About-Jaudet
2, and Noël Collignon
2.
Checked by Andrzej R. Daniewski, Bryon K. Tilley, and David L. Coffen.
1. Procedure
A. Bis(trifluoroethyl) methylphosphonate 1. All glassware is oven-dried. A 1-L, four-necked, round-bottomed flask is fitted with an efficient mechanical stirrer, a thermometer, reflux condenser with a bubbler, and a 200-mL, pressure-equalizing dropping funnel with a nitrogen inlet. Under a gentle flow of nitrogen, the flask is charged with 300 mL of anhydrous tetrahydrofuran (THF) (Note 1), 42 g (0.42 mol) of trifluoroethanol (Note 2), and 42.4 g of triethylamine (Note 2). Stirring is started, and the flask is immersed in a cool (~ 10°C) water bath. The dropping funnel is charged with a solution of 26.6 g (0.2 mol) of methylphosphonic dichloride (Note 3) in 100 mL of THF, that is subsequently added at a constant rate over 30 min. The internal temperature rises to 20–30°C, and a white solid precipitates. After the addition is complete, the water bath is removed, and the resulting mixture is stirred at room temperature for 2 hr. The organic salts are removed by suction filtration through a glass funnel, and the filter cake is washed with three 50-mL portions of THF. The solvent is completely removed under reduced pressure on a rotary evaporator. The residue is dissolved in 100 mL of dry ether and filtered to remove insoluble triethylamine hydrochloride, using an additional 40 mL of ether to aid the filtration/transfer. Solvent is again completely evaporated under reduced pressure. Crude product 1, thus obtained, is transferred to a 100-mL pear-shaped flask, fitted with a short distillation column (Note 4), and distilled under reduced pressure to afford 45.3–48.0 g (89–93%) of pure bis(trifluoroethyl) methylphosphonate (1) as a colorless liquid, bp 88–91°C (14 mm), which affords white crystals on standing in the freezer (Note 5). When sufficiently pure, 1 will crystallize on standing at room temperature.
B.
Bis(trifluoroethyl) (carboethoxymethyl)phosphonate (
2). An oven-dried, 1-L, four-necked, round-bottomed flask is fitted as above (Part A), flushed with
nitrogen, and charged with
131 mL (0.210 mol) of a solution of butyllithium (1.6 M in hexane) (Note 6). The solution is cooled with stirring to approximately −20°C in a
dry ice-acetone bath, and the dropping funnel is charged with a solution of
34 g (0.212 mol) of hexamethyldisilazane (Note 7) in
110 mL of tetrahydrofuran (THF) (Note 1). The contents of the addition funnel are added dropwise over 15 min during which time the yellow color practically disappears. A fine white suspension forms when the resulting solution is cooled to −78°C. A solution of
26 g (0.1 mol) of 1 and
11.5 g (0.106 mol) of ethyl chloroformate (Note 7) in
60 mL of THF is then added dropwise over 15 min via the addition funnel. During the course of the addition, the temperature rises to between −75° and −65°C, and the solution becomes clear. The reaction mixture is stirred at −70°C for an additional 15 min; the flask containing the reaction mixture is securely stoppered and stored overnight at −20°C in a freezer
(Note 8). The cold (−20°C) reaction mixture is poured into a stirred mixture of
170 mL of 2 M hydrochloric acid and an equal volume of crushed ice, and
150 mL of methylene chloride. The organic layer is separated, and the aqueous layer is extracted twice with
50 mL of methylene chloride (Note 9). The combined organic extracts are diluted with
200 mL of hexane to facilitate drying, and then dried for ~12 hr over anhydrous
magnesium sulfate (Note 10). After removal of the
magnesium sulfate by filtration, the solvents are evaporated under reduced pressure using a rotary evaporator to give crude
2 as a pale yellow liquid. Subsequent distillation of this crude material in a short path distillation apparatus
(Note 11) under reduced pressure gives, after a small forerun
(Note 12),
25.5–26.3 g (
77–79%) of
bis(trifluoroethyl) (carboethoxymethyl) phosphonate (2) as a colorless oil, bp
88–97°C (0.04 mm) (Note 13).
2. Notes
1.
Tetrahydrofuran available from SDS Company was purified by distillation from
sodium and
benzophenone.
2.
The submitters used
99+% 2,2,2-trifluoroethanol, and 99% triethylamine, purchased from Janssen Chimica, without purification. The checkers used the same grade materials purchased from Aldrich Chemical Company, Inc., and Eastman Kodak, respectively.
3.
Methylphosphonic dichloride (98%) can be purchased from Aldrich Chemical Company, Inc., or prepared according to a reported procedure.
34.
For the distillation, the submitters used a
14-cm fractional distillation column equipped with an
8-cm condenser.
5.
The product displays the following spectroscopic data:
31P (CDCl
3) δ: +35.0;
1H (CDCl
3) δ: 1.68 (d, 3 H, J = 18.2), 4.35–4.42 (m, 4 H);
13C (CDCl3) δ: 10.9, 61.8, 122.6; IR (film) cm
−1: 1415, 1317, 1292, 1258, 1186, 1169, 1111, 1075, 966, 922, 901, 845.
6.
The submitters used
140 mL of 1.5 M butyllithium in hexane available from Janssen Chimica, standardized before use by titration against a solution of
benzyl alcohol in
toluene and
2,2'-biquinoline. The checkers used material from Aldrich Chemical Company, Inc.
7.
The submitters used
98% 1,1,1,3,3,3-hexamethyldisilazane and 99% ethyl chloroformate purchased from Janssen Chimica and used without purification.
8.
Yields are low and erratic if the overnight cold storage is omitted!9.
Prolonged exposure to work-up conditions should be avoided to prevent hydrolysis at
phosphorus at this step.
10.
In some cases, the organic extracts are contaminated with a salt resulting from partial hydrolysis at
phosphorus. The salt precipitates slowly overnight and is removed together with the
magnesium sulfate by filtration prior to the removal of the solvents.
11.
A Büchi GKR-50 Kugelrohr distillation apparatus with three flasks was used by the submitters. The flask containing the crude product was in the upper part of the
oven, and the collecting flask was just outside.
12.
The forerun consisted mainly of siloxanes and other by-products.
13.
The product displays the following NMR and physical data:
31P (CDCl
3) δ: +24.0;
1H (CDCl
3) δ: 1.29 (t, 3 H, J = 7.1), 3.14 (d, 2 H, J = 21.2), 4.21 (q, 2 H, J = 7.2), 4.45 (m, 4 H);
13C (CDCl
3) δ: 14.1, 34.2, 62.8, 62.5, 122.7, 164.7. Anal. Calcd for C
8H
11F
6O
5P: C, 28.93; H, 3.34; F, 34.32; P, 9.33. Found: C, 29.10; H, 3.49; F, 34.20; P, 9.16.
Handling and Disposal of Hazardous Chemicals
The procedures in this article are intended for use only by persons with prior training in experimental organic chemistry. All hazardous materials should be handled using the standard procedures for work with chemicals described in references such as "Prudent Practices in the Laboratory" (The National Academies Press, Washington, D.C., 2011 www.nap.edu). All chemical waste should be disposed of in accordance with local regulations. For general guidelines for the management of chemical waste, see Chapter 8 of Prudent Practices.
These procedures must be conducted at one's own risk. Organic Syntheses, Inc., its Editors, and its Board of Directors do not warrant or guarantee the safety of individuals using these procedures and hereby disclaim any liability for any injuries or damages claimed to have resulted from or related in any way to the procedures herein.
3. Discussion
Ethyl bis(trifluoroethyl) phosphonoacetate was introduced to synthesis by Still and Gennari in 1983.
4 It is a very useful reagent for the preparation of Z-acrylates by means of a Wittig-Hürner olefination. The procedure described here illustrates a general method for the one-pot formation of phosphonocarboxylates and enolates. The key step in the sequence is the formation of an α-lithiomethyl phosphonate in the presence of two equivalents of a hindered lithium amide, which promotes the direct generation of the enolate.
5 The present procedure has several advantages: 1) easy availability of reagents, 2) the possibility to extend the reaction to various carboxylate groups by the choice of the chloroformates (e.g., ClCO
2R
3; R
3 = Me, Et, i-Pr, tert-Bu), 3) the possibility to extend the reaction to α-alkylated phosphonocarboxylates by the choice of the starting alkylphosphonate (R
1O)
2P(O)CH
2R
2 (R
2 = H, Me, Et, Pr, etc.), and 4) high yields of the phosphonates, simplicity, and ease of isolation of the products.
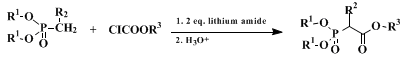
Appendix
Chemical Abstracts Nomenclature (Collective Index Number);
(Registry Number)
hydrochloric acid (7647-01-0)
ether (60-29-7)
PHOSPHORUS (7723-14-0)
nitrogen (7727-37-9)
toluene (108-88-3)
Benzophenone (119-61-9)
sodium (13966-32-0)
Benzyl alcohol (100-51-6)
Triethylamine hydrochloride (554-68-7)
methylene chloride (75-09-2)
ethyl chloroformate (541-41-3)
magnesium sulfate (7487-88-9)
butyllithium (109-72-8)
Tetrahydrofuran,
THF (109-99-9)
hexane (110-54-3)
triethylamine (121-44-8)
hexamethyldisilazane (999-97-3)
Bis(trifluoroethyl) (carboethoxymethyl)phosphonate,
Acetic acid, [bis(2,2,2-trifluoroethoxy)phosphinyl]-, ethyl ester,
bis(trifluoroethyl) (carboethoxymethyl) phosphonate (124755-24-4)
Bis(trifluoroethyl) methylphosphonate
trifluoroethanol (75-89-8)
methylphosphonic dichloride (3279-26-3)
2,2,2-trifluoroethanol (75-89-8)
2,2'-biquinoline (119-91-5)
1,1,1,3,3,3-hexamethyldisilazane (999-97-3)
Ethyl bis(trifluoroethyl) phosphonoacetate
Copyright © 1921-, Organic Syntheses, Inc. All Rights Reserved