Org. Synth. 1996, 73, 1
DOI: 10.15227/orgsyn.073.0001
3-[(1S)-1,2-DIHYDROXYETHYL]-1,5-DIHYDRO-3H-2,4-BENZODIOXEPINE
[1,2-Ethanediol, 1-(1,5-dihydro-2,4-benzodioxepin-3-yl)-, (S)-]
Submitted by Ryu Oi
1 and K. Barry Sharpless
2.
Checked by Michelle A. Laci and Robert K. Boeckman, Jr..
1. Procedure
CAUTION: Lithium aluminum hydride is a flammable solid which evolves hydrogen, a highly flammable and potentially explosive gas, upon contact with moisture. Potassium osmate is highly toxic as are any residues containing osmium and should be handled with care. All operations should be conducted in an efficient fume hood.
A. 1,2-Benzenedimethanol. An oven-dried, 2-L, three-necked, round-bottomed flask, equipped for mechanical stirring, and outfitted with a reflux condenser bearing an argon inlet/outlet vented through a mineral oil bubbler, and a 250-mL, pressure-equalizing addition funnel, is placed under an argon atmosphere and charged with 20.94 g (0.552 mol) of lithium aluminum hydride (LiAlH4), and 850 mL of anhydrous diethyl ether (Note 1) and (Note 2). A solution of 87.46 mL (97.78 g, 0.44 mol) of diethyl phthalate (Note 1) in 170 mL of anhydrous tetrahydrofuran (THF) is added dropwise via the addition funnel over 1.5 hr with efficient mechanical stirring, at a rate of addition adjusted to maintain a gentle reflux (Note 3). After the addition is complete, the mixture is heated to reflux by means of a heating mantle for 1.5 hr. The mechanically stirred reaction mixture is then cooled to ~0°C in an ice-water bath, and quenched by cautious sequential addition of 21 mL of water, 21 mL of aqueous 15% sodium hydroxide solution, and 63 mL of water (Note 4). While stirring is continued, the mixture is held for 30 min at 0°C then allowed to warm to room temperature. The resulting white solids are collected by vacuum filtration and washed on the filter using a total of 1 L of ether in 100-mL portions (Note 5). Concentration of the filtrate, under reduced pressure, affords 56.64 g (93%) of 1,2-benzenedimethanol as a white solid, mp 62–64°C, which is sufficiently pure for further transformation (Note 6).
B. 3-Vinyl-1,5-dihydro-3H-2,4-benzodioxepine. An oven-dried, 2-L, three-necked, round-bottomed flask is equipped with a magnetic stirring bar, a calcium chloride drying tube, and two rubber septa. The flask is charged with 41.45 g (0.3 mol) of 1,2-benzenedimethanol, 5.71 g (0.03 mol) of p-toluenesulfonic acid monohydrate (Note 1), and 600 mL of dry 1,2-dimethoxyethane (Note 1) and (Note 7). Stirring is initiated, and 49.2 mL (0.45 mol) of trimethyl orthoformate (Note 1) and 30.1 mL (0.45 mol) of acrolein (Note 1) are then added sequentially via syringe. The reaction mixture is stirred at room temperature for 5 hr (Note 8), then poured into a mixture of ice and 150 mL of aqueous 10% sodium bicarbonate solution in a 2-L separatory funnel using 90 mL of ether to rinse the flask. After thorough mixing, the organic phase is separated and the aqueous phase is extracted with 300 mL of ether. The combined organic layers are washed sequentially with 90 mL of cold aqueous 10% sodium bicarbonate solution and 90 mL of brine, dried over anhydrous sodium sulfate (Na2SO4), filtered, and concentrated under reduced pressure to give 52 g (99%) of crude acetal. Distillation of the residual liquid under reduced pressure affords 39.6–40.4 g (75–77%) of 3-vinyl-1,5-dihydro-3H-2,4-benzodioxepine as a colorless liquid, bp 116–118°C (5 mm), that may crystallize on standing, mp ~30°C (Note 9) and (Note 10).
C. 3-[(1S)-1,2-Dihydroxyethyl)]-1,5-dihydro-3H-2,4-benzodioxepine. A 2-L, three-necked, round-bottomed flask is equipped with a mechanical stirrer, a thermometer, and a rubber septum. The flask is charged with dihydroquinidine 9-O-(9'-phenanthryl) ether (Note 1) and (Note 11), [0.75 g, (1.5 mmol, 1 mol%, 0.0057 M in the organic phase)], 148.2 g (0.45 mol) of potassium ferricyanide (Note 1), 62.2 g (0.45 mol) of potassium carbonate (Note 1), 264 mL of tert-butyl alcohol (tert-BuOH) (Note 1), and 264 mL of distilled water. Mechanical stirring is initiated, 0.11 g (0.3 mmol, 0.2 mol%) of potassium osmate (VI) dihydrate, which generates a 0.0011 M solution of OsO4 in the organic phase (Note 1), is added, and the mixture is stirred at room temperature for 0.5 hr, resulting in an orange suspension. The flask is immersed in a 0°C cooling bath, and the mixture stirred for 1 hr (Note 12). Over a period of 24 hr at 0°C, 26.25 g (0.16 mol) of 3-vinyl-1,5-dihydro-3H-2,4-benzodioxepine is added dropwise via syringe (Note 13), and the reaction mixture is stirred an additional 24 hr at 0°C (Note 14). After the required time period has elapsed, 30.25 g (0.24 mol) of solid sodium sulfite (Na2SO3) is added in portions, the reaction mixture is stirred for an additional 1 hr at room temperature, poured into a 3-L beaker, and allowed to stand for another 1 hr at room temperature (Note 1). After the organic phase is decanted, the aqueous phase is diluted with 300 mL of water to dissolve the salts, and poured into a 2-L separatory funnel using 90 mL of dichloromethane (CH2Cl2) to rinse the flask and beaker. The aqueous phase is extracted three times with 150 mL of CH2Cl2. The organic extracts are combined with the decanted organic phase, dried over Na2SO4, filtered, and concentrated, under reduced pressure, to afford 33.1 g (104%) of crude product, as a pale yellow liquid (Note 15) that crystallizes rapidly. The ee of the crude 3-[(1S)-1,2-dihydroxyethyl)]-1,5-dihydro-3H-2,4-benzodioxepine is determined by HPLC analysis of the derived bis-Mosher ester to be 84% (Note 16).
A 2-L Erlenmeyer flask is charged with 33.1 g of the crude protected diol which is dissolved in 600 mL of hot ethyl acetate (EtOAc), and the solution is allowed to stand at room temperature for 3 hr and at 0°C to −5°C for 24 hr (Note 17). The resulting white precipitate is collected by suction filtration through a 10-cm Büchner funnel (Note 18), and the mother liquor is concentrated under reduced pressure to afford 21.15 g of a pale yellow liquid that slowly crystallizes (Note 19). The residual liquid is placed in a 500-mL Erlenmeyer flask, dissolved in 150 mL of hot toluene (Note 20), and allowed to stand for 24 hr at 0°C to −5°C. The slightly yellow precipitate is collected by suction filtration through a 10-cm Büchner funnel and dried under reduced pressure to give 16.0–17.4 g (50–55%) of 3-[(1S)-1,2-dihydroxyethyl)]-1,5-dihydro-3H-2,4-benzodioxepine, mp 73–75°C, [α]D23 −12.4° (CHCl3, c 2.62) (Note 21). The ee of the product is determined by HPLC analysis of the derived bis-Mosher ester to be 97% (Note 16).
The mother liquor is then concentrated to afford 2.4 g of a brown yellow liquid (Note 22). Flash chromatography of this material on 30 g of SiO2 with elution by EtOAc then by EtOAc:MeOH (5:1 v/v) affords 0.63 g (2%) of 3-[(1S)-1,2-dihydroxyethyl)]-1,5-dihydro-3H-2,4-benzodioxepine and 0.69 g (92% recovery) of dihydroquinidine 9-O-(9'-phenanthryl) ether.
2. Notes
1.
This reagent was purchased from Aldrich Chemical Company, Inc., and used without further purification.
2.
Anhydrous ether was purchased from J. T. Baker Chemical Company and used as received.
3.
Tetrahydrofuran was purchased from J. T. Baker Chemical Company and dried and deoxygenated prior to use by distillation from
sodium-benzophenone ketyl under
argon.
4.
During the initial stages of the addition of water, care must be exercised to control the vigorous exothermic reaction by use of efficient cooling and agitation along with careful regulation of the rate of addition. This procedure must be conducted in an efficient fume hood since a considerable volume of
hydrogen gas is produced that must be vented.
5.
Since the solids have a tendency to occlude significant amounts of the diol, efficient washing of the solids is essential to obtain good yields. Warm solvent can be employed to assist in removal of the product.
6.
1,2-Benzenedimethanol (lit.
3 mp
63–65°C) has the following spectral characteristics:
1H NMR (300 MHz, CDCl
3) δ: 3.97 (s, 2 H), 4.61 (s, 4 H), 7.30 (s, 4 H);
13C NMR (75 MHz, CDCl
3) δ: 63.8, 128.5, 129.6, 139.3; IR (CHCl
3) cm
−1: 3279, 2890, 1454, 1214, 1182, 1110, 1036, 1005, 758.
7.
A freshly opened bottle of
reagent grade 1,2-dimethoxyethane (Aldrich Chemical Company, Inc.) was used without further purification.
8.
The reaction mixture was monitored by TLC analysis on
silica gel (
4:1 EtOAc:hexane v/v).
3-Methoxy-1,5-dihydro-3H-2,4-benzodioxepine (R
f = 0.5) is first produced by transesterification with
trimethyl orthoformate, and is then transformed to the final product (R
f = 0.6).
9.
Slightly impure forerun, 6–9 g, was separated from the main fraction. After the distillation was stopped, approximately 6–9 g of residue remained.
10.
The spectroscopic data and elemental analysis of the product are as follows:
1H NMR (300 MHz, CDCl
3) δ: 4.91 (q, 4 H, J = 14.2), 5.33–5.38 (m, 2 H), 5.53 (dt, 1 H, J = 17.4, 1.4), 5.90–5.99 (m, 1 H), 7.10–7.25 (m, 4 H);
13C NMR (75 MHz, CDCl
3) δ: 70.0, 104.2, 118.4, 127.0, 127.2, 134.7, 138.8; IR (neat) cm
−1: 3068, 3024, 2950, 2860, 1769, 1725, 1497, 1445, 1412, 1376, 1353, 1291, 1268, 1221, 1210, 1150, 1123, 1094, 1046, 1032, 944, 932, 749; m/z Calcd. for Cs
+-C
11H
12O
2: 308.9892; Found 308.9892; Anal. Calcd for C
11H
12O
2: C, 74.98; H, 6.86; O, 18.16. Found: C, 75.12; H, 6.84; O, 18.26.
11.
Dihydroquinidine 9-O-(9'-phenanthryl) ether4 (Aldrich, Cat. No. 38195-0: Hydroquinidine 9-phenanthryl ether) gave the highest enantioselectivity compared with several other commercially available dihydroquinidine derivatives; 34% ee with
dihydroquinidine 4-chlorobenzoate (Aldrich, Cat. No. 33648-3: Hydroquinidine 4-chlorobenzoate) and 61% ee with
dihydroquinidine-9-O-(4'-methyl-2'-quinolyl) ether (Aldrich, Cat. No. 38194-2: Hydroquinidine 4-methyl-2-quinolyl ether). The submitters report that the reaction can be run on 0.5-mol scale using this procedure to afford comparable yields and enantiomeric purity of the diol product.
12.
Upon cooling, the mixture becomes a viscous suspension, and vigorous stirring is required. At temperatures below −5°C, the reaction mixture starts to freeze, thereby precluding efficient mixing.
13.
Neat
3-vinyl-1,5-dihydro-3H-2,4-benzodioxepine was added by syringe drive (Syringe Infusion Pump 22, Harvard Apparatus, South Natick, MA) using a gas-tight syringe. If the material has crystallized, it is liquified by gentle warming (~30°C) and
2 mL of t-BuOH-H2O is added before placing the material in the syringe. The tip of the syringe needle is 15 cm above the reaction surface to prevent freezing of the liquid which occurs around 5°C. Careful temperature control is required to prevent the mixture from freezing during the slow addition. Without slow addition, the ee of the product drops to 67%. The ee also deteriorates if the mixture is allowed to freeze during addition, permitting unreacted olefin to accumulate prior to warming enough to achieve efficient stirring.
14.
Reaction progress was monitored by TLC on
silica gel (
5:1 EtOAc:MeOH v/v). As the reaction neared completion, the color of the reaction mixture turned yellow.
15.
The oil contained 5–10%
t-BuOH by weight.
3-[(1R)-1,2-Dihydroxyethyl)]-1,5-dihydro-3H-2,4-benzodioxepine was produced with 84% ee when
dihydroquinine-9-O-(9'-phenanthryl) ether (Aldrich, Cat. No. 38197-7: Hydroquinine 9-phenanthryl ether) (Note 1) was used instead of
dihydroquinidine 9-O-(9'-phenanthryl) ether.
516.
The ee was determined by HPLC analysis (Chemcosorb 3Si, Chemco, Japan) of the corresponding bis-Mosher
6 ester eluted with
5% EtOAc in hexane (2 mL/min, (R)-diol: t
R = 14.8 min, (S)-diol: t
R = 18.2 min). The checkers employed a
25 × 10-mm Prep Nova-Pak HR Silica Column (particle size 6 mm, 60 Å) with UV detection at 220 nm and elution by
10% diethyl ether/hexane at 12 mL per min flow rate (R-diol: t
R = 12.6 min and S-diol: t
R = 15.1 min).
17.
Ethyl acetate is preferable to aromatic hydrocarbon solvents such as
benzene or
toluene because of the high solubility of the diol in
EtOAc; this results in higher recovery of the enantomerically enriched diol from the mother liquor.
18.
The white precipitate was dried under reduced pressure to afford
8.7–11.1 g (
28–35%) of
3-[(1S)-1,2-dihydroxyethyl)]-1,5-dihydro-3H-2,4-benzodioxepine (55–60% ee).
19.
The ee of this crude mixture before recrystallization was 97%.
20.
Aromatic hydrocarbon solvents such as
toluene or
benzene are preferable to several other solvents such as
EtOAc or
ethanol because of efficient recovery of the diol and better separation of the diol from the ligand.
21.
The melting point of the racemic diol was
110–113°C. The spectral data and elemental analysis of the diol are as follows:
1H NMR (300 MHz, CDCl
3) δ: 2.13 (t, 1 H, J = 6.4), 2.57 (d, 1 H, J = 4.2), 3.73 (m, 1 H), 3.80 (dd, 2 H, J = 6.4, 4.2), 4.93–5.01 (m, 5 H), 7.20–7 30 (m, 4 H);
13C NMR (75 MHz, CDCl
3) δ: 62.42, 72.17, 72.86, 73.23, 108.20, 127.73, 127.79, 138.89, 138.93; IR (CHCl
3) cm
−1: 3575, 3018, 2966, 2892, 2855, 1456, 1446, 1375, 1294, 1265, 1138, 1103, 1083, 1048; m/z Calcd. for Na
+-C
11H
14O
4: 233.0790; Found 233.0970; Anal. Calcd for C
11H
14O
4: C, 62.85; H, 6.71; Found: C, 62.78; H, 6.57.
22.
The oil contained 10–30%
toluene by weight.
Handling and Disposal of Hazardous Chemicals
The procedures in this article are intended for use only by persons with prior training in experimental organic chemistry. All hazardous materials should be handled using the standard procedures for work with chemicals described in references such as "Prudent Practices in the Laboratory" (The National Academies Press, Washington, D.C., 2011 www.nap.edu). All chemical waste should be disposed of in accordance with local regulations. For general guidelines for the management of chemical waste, see Chapter 8 of Prudent Practices.
These procedures must be conducted at one's own risk. Organic Syntheses, Inc., its Editors, and its Board of Directors do not warrant or guarantee the safety of individuals using these procedures and hereby disclaim any liability for any injuries or damages claimed to have resulted from or related in any way to the procedures herein.
3. Discussion
This procedure describes a convenient preparation of highly enantiomerically enriched
3-(1,2-dihydroxyethyl)-1,5-dihydro-3H,2,4-benzodioxepine,
5 a chiral
glyceraldehyde equivalent, on a 0.5-mol scale and illustrates the utility of catalytic asymmetric dihydroxylation (ADH)
7 on a large scale. The merits of the diol compared with other chiral derivatives of
glyceraldehyde, e.g.,
2,3-O-isopropylidene-, 2,3-O-cyclohexylidene-, or 2,3-di-O-acylated glyceraldehyde8 9 include its ease of handling, its low volatility and high stability at ambient temperature, and its UV chromophore that facilitates TLC or HPLC analysis. In addition, this seven-membered ring acetal can be deprotected easily under mild, neutral conditions by catalytic hydrogenolysis.
5,10 11 A variety of selective nucleophilic substitution reactions on derivatives easily obtained from this new C-3 chiral building block can be envisioned.
5The ADH at higher substrate concentrations is more practical for large scale applications. Unfortunately, high concentration of olefin in the reaction mixture is detrimental to the catalytic cycle responsible for asymmetric induction as the ee dropped to 67% at high olefin concentration (0.57 M in
t-BuOH) compared with 86% at low concentrations (0.13 M in
t-BuOH). However, in the more concentrated case, slow addition of the olefin raised the ee from 67% to 84%. The mechanistic rationale for the deleterious effect of high olefin concentration in this Os/K
3Fe(CN)
6 system
12 13 is not yet clear, but in any case the slow addition used here keeps the olefin concentration low.
Of the three types of racemates,
14 conglomerate, racemic compound, and solid solution,
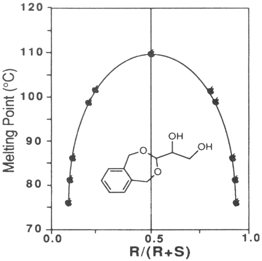
3-(1,2-dihydroxyethyl)-1,5-dihydro-3H-2,4-benzodioxepine shows melting point behavior characteristic of a racemic compound. The racemic diol is much higher melting than the enantiomerically enriched diol as shown in the
Figure 1. Therefore the diol of lower ee precipitates first during recrystallization and the enantiomerically enriched diol remains in the mother liquor. High ee diol (97% ee) is then obtained upon recrystallization of this mother liquor.
Figure 1 Relationship between enantiopurity and melting point.
Appendix
Chemical Abstracts Nomenclature (Collective Index Number);
(Registry Number)
silica gel
brine
SiO2
3-[(1S)-1,2-Dihydroxyethyl)]-1,5-dihydro-3H-2,4-benzodioxepine
potassium osmate
dihydroquinidine 9-O-(9'-phenanthryl) ether
ethyl acetate (EtOAc)
sodium-benzophenone ketyl
dihydroquinidine-9-O-(4'-methyl-2'-quinolyl) ether
t-BuOH-H2O
t-BuOH
3-[(1R)-1,2-Dihydroxyethyl)]-1,5-dihydro-3H-2,4-benzodioxepine
dihydroquinine-9-O-(9'-phenanthryl) ether
2,3-O-isopropylidene-, 2,3-O-cyclohexylidene-, or 2,3-di-O-acylated glyceraldehyde
ethanol (64-17-5)
potassium carbonate (584-08-7)
Benzene (71-43-2)
ethyl acetate,
EtOAc (141-78-6)
MeOH (67-56-1)
ether,
diethyl ether (60-29-7)
hydrogen (1333-74-0)
sodium sulfite (7757-83-7)
sodium hydroxide (1310-73-2)
Acrolein (107-02-8)
sodium bicarbonate (144-55-8)
sodium sulfate,
Na2SO4 (7757-82-6)
toluene (108-88-3)
dichloromethane,
CH2Cl2 (75-09-2)
Acrolein acetal
potassium ferricyanide (13746-66-2)
glyceraldehyde (56-82-6)
Tetrahydrofuran (109-99-9)
lithium aluminum hydride (16853-85-3)
hexane (110-54-3)
diethyl phthalate (84-66-2)
argon (7440-37-1)
tert-butyl alcohol (75-65-0)
1,2-dimethoxyethane (110-71-4)
osmium
trimethyl orthoformate (149-73-5)
glycidol (556-52-5)
p-toluenesulfonic acid monohydrate (6192-52-5)
3-[(1S)-1,2-Dihydroxyethyl]-1,5-dihydro-3H-2,4-benzodioxepine,
1,2-Ethanediol, 1-(1,5-dihydro-2,4-benzodioxepin-3-yl)-, (S)- (142235-22-1)
1,2-Benzenedimethanol (612-14-6)
3-Vinyl-1,5-dihydro-3H-2,4-benzodioxepine (142169-23-1)
3-Methoxy-1,5-dihydro-3H-2,4-benzodioxepine (67461-24-9)
dihydroquinidine 4-chlorobenzoate,
Hydroquinidine 4-chlorobenzoate
Hydroquinine 9-phenanthryl ether
3-(1,2-dihydroxyethyl)-1,5-dihydro-3H,2,4-benzodioxepine,
3-(1,2-dihydroxyethyl)-1,5-dihydro-3H-2,4-benzodioxepine
phenanthryl ether
Hydroquinidine 9-phenanthryl ether
Hydroquinidine 4-methyl-2-quinolyl ether
potassium osmate (VI) dihydrate (19718-36-3)
Copyright © 1921-, Organic Syntheses, Inc. All Rights Reserved