Org. Synth. 1992, 70, 139
DOI: 10.15227/orgsyn.070.0139
TETRAHYDRO-3-BENZAZEPIN-2-ONES BY LEAD TETRAACETATE OXIDATION OF ISOQUINOLINE ENAMIDES: 7,8-DIMETHOXY-1,3,4,5-TETRAHYDRO-2H-3-BENZAZEPIN-2-ONE
[2H-3-Benzazepin-2-one, 1,3,4,5-tetrahydro-7,8-dimethoxy-]
Submitted by George R. Lenz and Ralph A. Lessor
1.
Checked by Shaowo Liang and Leo A. Paquette.
1. Procedure
A. N-(tert-Butoxycarbonyl)-6,7-dimethoxy-1-methylene-1,2,3,4-tetrahydroisoquinoline. A 1-L, three-necked, round-bottomed flask is equipped with a thermometer, magnetic stirring bar, nitrogen inlet with gas bubbler, and a pressure-equalizing dropping funnel (Note 1). The flask is charged with 102.6 g (500 mmol) of 6,7-dimethoxy-1-methyl-3,4-dihydroisoquinoline (Note 2) and 150 mL of alcohol-free chloroform (Note 3). The mixture is warmed to 50°C with stirring. The dropping funnel is charged with a solution of 136.9 g (627 mmol) of di-tert-butyl pyrocarbonate (Note 4) in 50 mL of alcohol-free chloroform. The nitrogen is turned off, and the solution of di-tert-butyl pyrocarbonate is added to the mixture at such a rate as to maintain steady but controlled gas evolution (Note 5). The stirred reaction mixture is heated at 60–65°C until gas evolution ceases, then allowed to cool to room temperature with stirring.
Solvent is removed on a rotary evaporator at reduced pressure, and the resulting crude pink solid is dried overnight under high vacuum. The yield of slightly pink solid is 176.5–180 g (Note 6), (Note 7).
B. N-(tert-Butoxycarbonyl)-7,8-dimethoxy-1,3,4,5-tetrahydro-2H-3-benzazepin-2-one A 1-L, three-necked, round-bottomed flask is equipped with a nitrogen inlet, mechanical stirrer, thermometer, and a pressure-equalizing dropping funnel (Note 8). The flask is charged with 93 g (210 mmol) of lead tetraacetate (Note 9) and 250 mL of glacial acetic acid. Stirring is started, the flask is immersed in an ice-water bath, and the funnel is charged with a solution of 61 g (200 mmol) of N-(tert-butoxycarbonyl)-6,7-dimethoxy-1-methylene-1,2,3,4-tetrahydroisoquinoline (Note 10) in 250 mL of methylene chloride. This solution is added at such a rate that a temperature of 19–23°C is maintained throughout the addition, typically over a period of 15 to 20 min. The cooling bath is removed, and the mixture is stirred at room temperature for 1 hr. Glycerol (4 mL) is added to quench unreacted lead tetraacetate, and the mixture is stirred for an additional 10 min.
The mixture is poured into 750 mL of water in a 2-L separatory funnel and shaken thoroughly. The phases are separated, and the aqueous phase is extracted with two 100-mL portions of methylene chloride. The combined organic layers are washed with 700 mL of water, followed by successive 100-mL portions of saturated aqueous sodium bicarbonate until no further effervescence is observed. The organic layer is dried over magnesium sulfate, filtered, and evaporated at reduced pressure to give a yellow-orange solid, which is dissolved in 100 mL of boiling acetone and allowed to cool slowly to room temperature, then kept overnight at −20°C. Filtration affords a cream-colored solid in a yield of 51.2–52.2 g (Note 11), (Note 12). A second crop is obtained by evaporation of the mother liquors, dissolution in a minimal amount of boiling acetone, cooling and seeding. The total yield is brought to 58–59 g (90–92%).
C. 7,8-Dimethoxy-1,3,4,5-tetrahydro-2H-3-benzazepin-2-one. A 250-mL, three-necked, round-bottomed flask is equipped with a magnetic stirring bar, thermometer, and pressure-equalizing dropping funnel. The flask is charged with 32.1 g (100 mmol) of recrystallized N-(tert-butoxycarbonyl)-7,8-dimethoxy-1,3,4,5-tetrahydro-2H-3-benzazepin-2-one and 100 mL of methylene chloride. The flask is immersed in an ice-water bath and stirred until the internal temperature reaches 5°C. The dropping funnel is charged with 40 mL of trifluoroacetic acid, which is added dropwise at a rate such that the temperature of the reaction mixture does not exceed 10°C (Note 13). When addition is complete, the cooling bath is removed, and the mixture is stirred for 3 hr (Note 14).
The mixture is diluted with an additional 700 mL of methylene chloride and poured into a separatory funnel containing 500 mL of water. The funnel is shaken thoroughly, and the phases are separated. The organic layer is washed with 500 mL of water, followed by successive washes with 200-mL portions of saturated aqueous sodium bicarbonate until no further effervescence is observed (Note 15). The organic phase is dried over sodium sulfate and evaporated under reduced pressure to give 24.8–25.2 g of crude product. The crude material is dissolved in a minimal amount of boiling methylene chloride, and then diluted with an equal volume of ethyl acetate. The mixture is boiled down to approximately two thirds of its starting volume, by which time crystallization has begun, then allowed to cool slowly to room temperature. After storage at −20°C overnight, filtration affords 17.5–18.0 g (79–81%) of product, mp 194–195°C (Note 16), (Note 17)).
2. Notes
1.
The glassware is dried in an
oven at 110°C and assembled while still hot, then allowed to cool while a slow stream of
nitrogen is passed through the apparatus.
2.
6,7-Dimethoxy-1-methyl-3,4-dihydroisoquinoline was prepared according to an
Organic Syntheses procedure: Brossi, A.; Dolan, L. A.; Teitel, S.
Org. Synth., Coll. Vol. VI 1988, 1.
3.
The submitters used
J. T. Baker Chemical Company hydrocarbon-stabilized chloroform containing 0.015% amylene stabilizer. The checkers used
chloroform of comparable quality purchased from Aldrich Chemical Company, Inc.4.
The submitters used commercially available
tert-butyl pyrocarbonate from either Fluka or Aldrich Chemical Company, Inc. Alternatively, the reagent can be prepared according to Pope, B. M.; Yamamoto, Y.; Tarbell, D. S.
Org. Synth., Coll. Vol. VI 1988, 418.
5.
The addition typically took 1.5 to 2 hr. The bubbler on the
nitrogen line used to flush the flask is conveniently used to monitor the evolution of
carbon dioxide as the reaction proceeds.
6.
This material is pure enough for use in the next step. The impurities of
tert-butyl alcohol and a small amount of unreacted
tert-butyl pyrocarbonate do not interfere with the oxidation.
7.
If desired, the material can be recrystallized from
methanol; under these circumstances,
146–149 g of white solid, mp
101–102°C, is returned. The spectral characteristics of recrystallized material are as follows:
1H NMR (300 MHz, CDCl
3) δ: 1.49 (s, 9 H), 2.78 (t, 2 H, J = 5.9), 3.77 (t, 2 H, J = 5.9), 3.86 (s, 3 H), 3.89 (s, 3 H), 5.31 (s, 1 H), 5.50 (s, 1 H), 6.56 (s, 1 H), 7.11 (s, 1 H); IR (KBr) cm
−1: 1690, 1630, 1605, 1510, 1390, and 1170;
13C NMR (75 MHz, CDCl
3) δ: 28.38, 28.96, 43.51, 55.89, 56.02, 80.47, 101.86, 107.46, 110.77, 124.84, 127.72, 139.99, 147.51, 149.22, 153.83. Anal. Calcd. for C
17H
23NO
4: C, 66.86; H, 7.59; N, 4.59. Found: C, 67.02; H, 7.48; N, 4.65.
8.
The glassware is assembled hot under
nitrogen as for the previous step. The nitrogen inlet and stirrer are mounted on a
Claisen adapter, and the thermometer is removed during the flushing period, then reinserted.
9.
The submitters used
lead tetraacetate from Aldrich Chemical Company, Inc., which was dried under reduced pressure at room temperature for 10 min prior to use to remove any
acetic acid present. Alternatively,
lead tetraacetate still containing
acetic acid may be used successfully if a slight excess is used.
10.
If crude material containing
tert-butyl alcohol and unreacted pyrocarbonate is used in this step, the amount of starting material present is calculated based on the mass balance for the first step, assuming a quantitative conversion, and 1.05 equivalents of
lead tetraacetate are used. The checkers used only pure material and advise against carrying forward less pure carbamate.
11.
This material may be used directly in the following step. If desired, the material can be recrystallized from
acetone, mp
116.5–118°C. The spectral characteristics of the recrystallized material are as follows:
1H NMR (300 MHz, CDCl
3) δ: 1.52 (s, 9 H), 3.14 (t, 2 H, J = 6.0), 3.84 (s, 6 H), 3.92 (s, 2 H), 4.18 (t, 2 H, J = 6.0), 6.56 (s, 1 H), 6.57 (s, 1 H); IR (KBr) cm
−1: 1715, 1610, 1525, 1370, 1255, 1110, and 1060;
13C NMR (75 MHz, CDCl
3) δ: 28.03, 32.83, 43.42, 45.21, 55.94, 55.97, 83.18, 113.25, 114.28, 121.92, 127.09, 147.41, 148.38, 152.08, 171.33. Anal. Calcd. for C
17H
23NO
5: C, 63.54; H, 7.21; N, 4.36. Found: C, 63.36; H, 7.30; N, 4.16.
12.
If insufficient
lead tetraacetate is used in the oxidation, unoxidized starting enamide is hydrolyzed during the workup to
tert-butyl 2-(2-acetyl-3,4-dimethoxyphenyl) ethyl carbamate, mp
111.5–112.5°C (cf. ref. in
(Note 2)):
1H NMR (270 MHz, CDCl
3) δ: 1.42 (s, 9 H), 2.58 (s, 3 H), 3.03 (t, 2 H), 3.37 (q, 2 H), 3.92 (s, 3 H), 6.76 (s, 1 H), 7.23 (s, 1 H); IR (FTIR) cm
−1: 1707, 1674, 1604, 1517, 1266, 1212, 1152. Anal. Calcd for C
17H
25NO
5: C, 63.13; H, 7.79; N, 4.33. Found: C, 63.24; H, 7.91; N, 4.28. The hydrolyzed material co-migrates with the oxidation product in a variety of TLC systems, and also co-crystallizes with it. It is, however, removed during the
trifluoroacetic acid (TFA) cleavage to form the benzazepinone (Step C). The presence of any hydrolyzed material is readily detected by the presence of the acetyl resonance (δ 2.58) in the NMR spectrum.
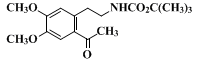
13.
After approximately
25 mL of the trifluoroacetic acid have been added, gas evolution begins. This can be quite vigorous if the temperature is not kept below 10°C.
14.
The progress of the reaction can be monitored by thin layer chromatography on silica gel plates, using a
95:5:0.5 mixture of chloroform:methanol:concentrated ammonium hydroxide as the developing solvent.
15.
Foaming can be quite vigorous, especially if the reaction mixture is not washed first with water prior to the use of
sodium bicarbonate solution.
16.
A small second crop of impure material can be obtained from the mother liquors.
17.
The product exhibited the following spectral characteristics:
1H NMR (300 MHz, CDCl
3) δ: 3.03 (t, 2 H, J = 6.0), 3.52–3.60 (m, 2 H), 3.75 (s, 2 H), 3.83 (s, 3 H), 3.84 (s, 3 H), 6.34 (br s, 1 H), 6.59 (s, 1 H), 6.62 (s, 1 H);
13C NMR (75 MHz, CDCl
3) δ: 33.03, 40.62, 41.71, 55.77 (2C), 112.96, 113.53, 123.19, 128.39, 147.09, 147.72, 174.49; IR (KBr) cm
−1: 1675, 1220, 1125, 1100, and 1010.
Handling and Disposal of Hazardous Chemicals
The procedures in this article are intended for use only by persons with prior training in experimental organic chemistry. All hazardous materials should be handled using the standard procedures for work with chemicals described in references such as "Prudent Practices in the Laboratory" (The National Academies Press, Washington, D.C., 2011 www.nap.edu). All chemical waste should be disposed of in accordance with local regulations. For general guidelines for the management of chemical waste, see Chapter 8 of Prudent Practices.
These procedures must be conducted at one's own risk. Organic Syntheses, Inc., its Editors, and its Board of Directors do not warrant or guarantee the safety of individuals using these procedures and hereby disclaim any liability for any injuries or damages claimed to have resulted from or related in any way to the procedures herein.
3. Discussion
This procedure illustrates a general route to tetrahydro-3-benzazepin-2-ones from readily available dihydroisoquinolines.
2 The benzazepine ring system exists in various classes of isoquinoline-derived alkaloids,
3 4 5 while other members of this class are being developed as pharmaceutical agents.
6 The present procedure takes advantage of ready formation of enamides from dihydroisoquinolines and carboxylic acid anhydrides, acid chlorides, carbonic anhydrides and chlorides and their facile oxidation to differentially functionalized benzazepinones (Table I). The mechanism has been described and involves migration of the isoquinoline aromatic ring to the exocyclic methylene group.
2
TABLE I
LEAD TETRAACETATE OXIDATIVE RING EXPANSION OF ISOQUINOLINE ENAMIDES
|
|
|
|
Yield (%)
|
|
|
|
62
|
|
|
91
|
|
|
92
|
|
|
94
|
|
|
50
|
|
|
72
|
|
Several approaches to the synthesis of the tetrahydrobenzazepine ring system have been described,
7 and excellent methods exist for the preparation of aryl substituted tetrahydrobenzazepines.
6 However, benzazepines that are either unsubstituted or alkyl-substituted on the azepine ring are much less readily obtainable. For instance, the benzazepinone, synthesized by this procedure, was originally isolated, in low yield, from the mixture of photoproducts obtained from the irradiation of
N-[3-(3,4-dimethoxyphenyl)]propyl chloroacetamide.
8 9 Preparative approaches to the benzazepinones have required multiple steps starting from an
N-phenylethylacetamide and involving chloromethylation, cyanide displacement, nitrile solvolysis, hydrolysis to the amino acid and cyclization.
10 11 The 1-alkyl derivatives are subsequently prepared by alkylation of the parent compound.
12 The current procedure reduces the preparation of the tetrahydrobenzazepinone ring system to two straightforward steps.
Ring expansion of the isoquinoline enamides is insensitive to the type of acyl functionality used to form the enamide.
13 The reaction occurs when the isoquinoline aromatic ring is unsubstituted, or contains electron releasing substituents. The reaction is sensitive, however, to the degree and type of substitution on the exocyclic methylene group. Oxidative ring expansion occurs when the double bond is either unsubstituted or monoalkyl substituted. Phenyl substitution yields differing products depending on a number of variables.
13 When the exocyclic double bond is disubstituted, oxidation with
lead tetraacetate proceeds readily, but does not lead to ring expansion.
14 The ring expansion reaction works equally well for the preparation of tetrahydrobenzazocinones from tetrahydrobenzazepine enamides (Table II).
15
TABLE II
LEAD TETRAACETATE OXIDATIVE RING EXPANSION OF BENZAZEPINE ENAMIDES
|
|
|
|
Yield(%)
|
|
|
|
76
|
|
|
83
|
|
|
73
|
|
|
78
|
|
The acid-catalyzed cleavage of the tert-butoxycarbonyl group is the best method to form the parent benzazepinone. Other methods used have been the
Pd/C hydrogenolysis of a benzyloxycarbonyl group,
2,15,16 and
zinc mediated reductive cleavage of trichloroethoxy and trichloro-tert-butoxy carbonyl groups.
15
Appendix
Chemical Abstracts Nomenclature (Collective Index Number);
(Registry Number)
trifluoroacetic acid (TFA)
Pd/C
acetic acid (64-19-7)
ethyl acetate (141-78-6)
methanol (67-56-1)
chloroform (67-66-3)
glycerol (56-81-5)
sodium bicarbonate (144-55-8)
sodium sulfate (7757-82-6)
nitrogen (7727-37-9)
carbon dioxide (124-38-9)
acetone (67-64-1)
zinc (7440-66-6)
ammonium hydroxide (1336-21-6)
methylene chloride (75-09-2)
magnesium sulfate (7487-88-9)
amylene
tert-butyl alcohol (75-65-0)
trifluoroacetic acid (76-05-1)
N-phenylethylacetamide (877-95-2)
6,7-Dimethoxy-1-methyl-3,4-dihydroisoquinoline (4721-98-6)
BOC
7,8-Dimethoxy-1,3,4,5-tetrahydro-2H-3-benzazepin-2-one,
2H-3-Benzazepin-2-one, 1,3,4,5-tetrahydro-7,8-dimethoxy- (20925-64-8)
N-(tert-Butoxycarbonyl)-6,7-dimethoxy-1-methylene-1,2,3,4-tetrahydroisoquinoline (82044-08-4)
di-tert-butyl pyrocarbonate (24424-99-5)
N-(tert-Butoxycarbonyl)-7,8-dimethoxy-1,3,4,5-tetrahydro-2H-3-benzazepin-2-one (146858-74-4)
tert-butyl pyrocarbonate
tert-butyl 2-(2-acetyl-3,4-dimethoxyphenyl) ethyl carbamate
N-[3-(3,4-dimethoxyphenyl)]propyl chloroacetamide
lead tetraacetate (546-67-8)
Copyright © 1921-, Organic Syntheses, Inc. All Rights Reserved