Org. Synth. 1996, 73, 25
DOI: 10.15227/orgsyn.073.0025
ENANTIOSELECTIVE HYDROLYSIS OF cis-3,5-DIACETOXYCYCLOPENTENE: (1R,4S)-(+)-4-HYDROXY-2-CYCLOPENTENYL ACETATE
[4-Cyclopentene-1,3-diol, monoacetate, (1R-cis)-]
Submitted by Donald R. Deardorff, Colin Q. Windham, and Chris L. Craney
1.
Checked by Renaud Beaudegnies and Leon Ghosez.
1. Procedure
A. cis-3,5-Diacetoxycyclopentene. A flame-dried, 100-mL, single-necked, round-bottomed flask is equipped with a serum cap, Teflon-coated magnetic stirring bar, and an 18-gauge needle attached to a dry source of argon vented through a mineral oil bubbler. The apparatus is flushed with argon and charged with 16.70 g (118 mmol) of (±)-cis-4-acetoxy-1-hydroxycyclopent-2-ene [(±)-1] (Note 1) and 20 mL of methylene chloride (Note 2). To this stirred solution is added 8.81 g (129 mmol) of imidazole in one portion (Note 3). When dissolution is complete, the reaction mixture is cooled to 0°C with the aid of an ice-water bath. Acetic anhydride (13.20 g, 12.20 mL, 129 mmol) (Note 4) is added dropwise over 5 min by means of an oven-dried syringe and needle. The cooling bath is removed and the reaction is allowed to stir at ambient temperature until judged complete by TLC analysis (Note 5). The contents of the flask are transferred to a 250-mL separatory funnel and diluted with 60 mL of reagent grade ethyl ether. The contents of the separatory funnel are washed with 60 mL of ice-cold 1 N hydrochloric acid which results in noticeable removal of all color from the organic phase. The acidic aqueous phase is back-extracted with two 50-mL portions of ether (Note 6). The combined organic phases are washed with a single 50-mL portion of saturated sodium bicarbonate solution followed by back-extraction of the aqueous phase with three 50-mL portions of ether (Note 6). The combined organic extracts are dried over anhydrous magnesium sulfate, filtered, and concentrated under rotary evaporation at 30 mm. The resulting viscous tan oil is distilled through a 10-cm Vigreux column to afford 20.9–21.1 g (96–98%) of cis-3,5-diacetoxycyclopentene (2) (Note 7) as a colorless liquid, bp 55–56°C at 0.1 mm.
B.
(1R,4S)-(+)-4-Hydroxy-2-cyclopentenyl acetate. A rigorously clean
1-L Erlenmeyer flask, equipped with a
2-in. long Teflon-coated stirring bar (Note 8), is charged with
320 mL of a 1.45 M sodium dihydrogen phosphate buffer concentrate
(Note 9). The solution is diluted to a final volume of 800 mL with the addition of 480 mL of glass-distilled water. To the gently stirred solution
(Note 10) is added
78 mg of sodium azide (Note 11) followed by
18.6 mg (200 units/mg) of lyophilized electric eel acetyl cholinesterase (EEAC) (Note 12). The enzyme completely dissolves within 5 min after which time
16.01 g (87.0 mmol) of cis-3,5-diacetoxycyclopentene is added in one portion. A few extra mL of distilled water are used to facilitate a quantitative transfer of the diacetate. Parafilm is placed over the mouth of the flask and the two-phase system
(Note 13) is allowed to stir at room temperature
(Note 14). The progress of the reaction is conveniently monitored by periodic TLC analysis. The reaction is terminated when only a trace of diacetate remains and the corresponding diol begins to appear
(Note 15). The aqueous phase is first extracted three times with
150-mL portions of ether followed by fifteen 150-mL extractions with a 1:1 mixture of
ether/ethyl acetate (Note 16). The organic extracts are combined, dried over anhydrous
magnesium sulfate, and concentrated by rotory evaporation at ~30 mm
(Note 17). The residue is passed through a 60-g plug of silica gel
(Note 18) with approximately
750 mL of ether. Removal of the solvent by rotary evaporation followed by exposure to
oil pump vacuum affords
11.32 g of nearly colorless material [mp
43–49°C (corrected)]. Vacuum distillation
(Note 19) of the product through a short-path apparatus at 64°C/0.060 mm furnishes
10.64–10.76 g (
86–87%, 96% e.e.) of colorless solid (+) hydroxy acetate
1 (mp
44.5–49.5°C (corrected),
[α]D23 +62.9° (
chloroform,
c 1.465))
(Note 20). A sample recrystallized from 1:1
pentane/ether (approximately 5 mL/g) provides high purity (≥99% e.e.), colorless needles of
1 (mp
50.7–51.3°C (corrected),
[α]D23 +73.8° (
chloroform,
c 1.25) [lit.
2 mp
49° − 51°C,
[α]D23 +68.0° (
chloroform,
c 1.64)]
(Note 21).
2. Notes
1.
(±)-cis-4-Hydroxy-2-cyclopentenyl acetate [(±)-
1] was prepared according to the method described in
Org. Synth.
3 This material must be recrystallized (1:1
ether-
pentane) before use to remove impurities (e.g., trans-monoacetate) that otherwise lead to reduced optical rotations in the final product [(+)-
1]. The submitters have found that material having a melting point of
37–39°C is of satisfactory purity. The racemic monoacetate is hygroscopic so the proper precautions should be taken to prevent exposure to moisture. The NMR spectrum is as follows:
1H NMR of (±)
1 (200 MHz, CDCl
3) δ: 1.64 (dt, 1 H, J = 14.7, 3.7), 2.02 (s, 3 H), 2.78 (overlapping dt, 1 H, J = 14.7, 7.5), 4.70 (overlapping s, 1 H and broad s), 5.45 (m, 1 H), 5.95 (m, 1H), 6.10 (m, 1H);
13C NMR of (+)
1 (50 MHz, CDCl
3) δ: 21.2, 40.5, 74.9, 77.0, 132.6, 138.5, 170.8.
2.
HPLC grade methylene chloride was purchased from Fisher Scientific Company and distilled from
calcium hydride through a Vigreux column prior to use.
3.
Imidazole (99% indicated purity) was purchased from Aldrich Chemical Company, Inc., and used without any further purification.
4.
Acetic anhydride (98% indicated purity) was purchased from Aldrich Chemical Company, Inc., and distilled from
phosphorus pentoxide through a Vigreux column prior to use.
5.
Thin layer chromatographic (TLC) analysis was performed on Baker Si250F precoated plates with
ethyl ether as the solvent. The approximate R
f values for the monoacetate and diacetate under these conditions are 0.44 and 0.72, respectively. The compounds were visualized using
p-anisaldehyde stain.
6.
This step is necessary to retrieve the slightly water-soluble product diester that migrates into the aqueous layer during the wash process. The meso-diester is easily detected in the water phase by TLC analysis (see
(Note 5)).
7.
cis-3,5-Diacetoxycyclopentene has the following spectral data:
1H NMR of
2 (200 MHz, CDCl
3) δ: 1.74 (dt, 1 H, J = 15, 3.85), 2.05 (s, 6 H), 2.87 (overlapping dt, 1 H, J = 15, 7.6), 5.55 (m, 2 H), 6.1 (s, 2 H);
13C NMR of
2 (50 MHz, CDCl
3) δ: 21.1, 37.1, 76.6, 134.6, 170.7.
8.
The stirring bar should be free of any slivers of magnetic material or heavy metals that could inactivate the enzyme.
9.
The buffer was prepared by dissolving
100 g of sodium dihydrogen phosphate monohydrate (Alfa Johnson Matthey) into 200 mL of glass-distilled water. The pH of the solution was adjusted to 6.9 with the addition of concentrated
sodium hydroxide solution (MCB Reagent) and verified with a pH meter recently calibrated between 4–7 pH units using Fisher standards. The buffer solution was diluted to a final volume of 500 mL (1.45 M) and stored in a
clean plastic bottle.
10.
The stirring rate was adjusted so as to minimize the size of the vortex, which would introduce air into the solution and jeopardize the enzyme.
11.
Sodium azide (Sigma Chemical Company) was used to protect the solution's components from microbial attack during the prolonged reaction period.
12.
Lyophilized electric eel acetyl cholinesterase (C-3389, EC 3.1.1.7) was purchased from Sigma Chemical Company and stored below 0°C.
13.
As the reaction nears completion, all the diacetate dissolves.
14.
The success of this reaction may be temperature dependent. Shortly after starting this reaction on a particularly hot day, the building air conditioning failed. The temperature of the reaction flask quickly reached 28.7°C. The optical rotation of the distilled product was 4° off and the melting point was lower than expected. In all reactions prior to or since that incident the reaction temperatures never climbed above 23°C and the results have been uniformly superior.
15.
Under these conditions, the reaction lasts 9–12 hr with a final pH of approximately 6.5. Nonspecific hydrolysis of
2 over this time accounts for less than 0.5% of the product as measured by control experiments. If cost is of little concern, the reaction time can be shortened dramatically by the addition of extra enzyme.
cis-3,5-Dihydroxycyclopentene has an R
f value in ether of approximately 0.1.
16.
HPLC grade ethyl acetate was purchased from J. T. Baker Inc. and distilled from
phosphorus pentoxide prior to use. Although the product,
(1R,4S)-(+)-4-hydroxy-2-cyclopentenyl acetate [(+)-1], is more soluble in
ethyl acetate, emulsion concerns dictated the use of pure ether for the first three extractions and then a 1:1
ether/
ethyl acetate mixture for all subsequent extractions. When emulsions did occur during the extraction process, the unemulsified portion of the aqueous phase was drained and a few mL of
brine solution was added to the emulsified phase. Two or three quick shakes alleviated the problem.
17.
The residual oil may crystallize during this process. If so, it will be necessary to dissolve the solid in about
10 mL of ether (with the aid of a hot air gun) prior to passing the material through the silica gel plug.
18.
Baker Analyzed Reagent silica gel 40-140 mesh was used in a 4.5-cm diameter column. This step removes the small amounts of diol present. TLC analysis was helpful in determining when all the monoacetate had been stripped from the plug.
19.
The receiving flask should be submerged in an ice-water bath since the product has a considerable vapor pressure under these conditions. It is wise to have a heat gun nearby in the event that the condensate begins to crystallize in the condenser.
20.
The enantiomeric excess was unambiguously determined via gas chromatographic separation of diastereomers obtained from derivatization of the chiral alcohol with Mosher's reagent,
4 (R)-(+)-α-methoxy-α-(trifluoromethyl)phenylacetic acid, (MTPA) 99% (Aldrich Chemical Company, Inc.). The checkers used the procedure of Noyori: Kitamura, M.; Tokunaga, M.; Ohkuma, T., Noyori, R.
Org. Synth., Coll. Vol. IX 1998, 592 for derivatization with Mosher's reagent. The MTPA esters derived from the (−)- and (+)-monoacetate enantiomers have retention times of 108 and 111.5 min, respectively, on a 25-m OV-101, crosslinked, methylsilicone capillary column operating at 150°C and flow rate of 22 cm/sec. Although less reliable than GC, the ratio of MTPA esters can be estimated via the use of high field (≥200 MHz) NMR in CDCl
3 solvent.
521.
One paper reports5 an optical rotation of
[α]D22 +75.0° (
chloroform,
c 1.16) for crystals of (+)-
1 melting at only
47.5–48°C. We find these data to be inconsistent with our own experience and with the absolute values of the reported rotations for optically pure (−)-
1. The polarimeter used in the submitter's analysis, a JACSO DIP-360, had been previously calibrated with NBS Standard Reference
Sucrose 17d in accordance with NBS guidelines.
Handling and Disposal of Hazardous Chemicals
The procedures in this article are intended for use only by persons with prior training in experimental organic chemistry. All hazardous materials should be handled using the standard procedures for work with chemicals described in references such as "Prudent Practices in the Laboratory" (The National Academies Press, Washington, D.C., 2011 www.nap.edu). All chemical waste should be disposed of in accordance with local regulations. For general guidelines for the management of chemical waste, see Chapter 8 of Prudent Practices.
These procedures must be conducted at one's own risk. Organic Syntheses, Inc., its Editors, and its Board of Directors do not warrant or guarantee the safety of individuals using these procedures and hereby disclaim any liability for any injuries or damages claimed to have resulted from or related in any way to the procedures herein.
3. Discussion
In the 1980's there was a great increase in the development and use of enzymatic procedures by synthetic chemists.
6,7,8,9,10,11 Previously regarded more as scientific curiosities of limited scope than of practical utility, biological-chemical transformations are now used regularly by synthetic chemists. The ability to induce optical activity in molecules where none existed before is the most useful property of these chiral catalysts. Hydrolase enzymes are generally preferred over other kinds of enzymes for transformations of this nature because they are more easily handled and do not require cofactors for activity. In cases where enantiotopic differentiation between ester functions is desired, prochiral meso diesters are more efficient substrates than racemic esters. In the former case it is possible for all starting material to be converted into a single enantiomer, whereas in the latter example only enzymatic resolution is possible.
(1R,4S)-(+)-4-Hydroxy-2-cyclopentenyl acetate [(+)-
1] is an important synthetic precursor. It provides optically active starting material, via the versatile intermediate
4-oxo-2-cyclopentenyl acetate,
12 for important cyclopentanoids such as the prostaglandins
13 and carbocyclic nucleosides.
14 Because of the medicinal significance of these compounds, more efficient routes with better enantioselectivities have been devised to nonracemic
1. Enzymatic catalysis has become the dominant methodology for induction of this optical activity.
In addition to the title procedure,
15 other enzymatic preparations of nearly optically pure
(1R,4S)-(+)-4-hydroxy-2-cyclopentenyl acetate [(+)-
1]
5,21 and its optical antipode (−)-
116,17,18,19,20 are known. These enzyme-catalyzed procedures are derivatives of two basic strategies: (1) the enantioselective hydrolysis of the meso-diacetate
2,
5,15,21,16,17 or (2) the enantioselective transacetylation of the parent meso-diol.
18,19,20 Although (−)-
1 has been successfully prepared by either route, the (+)-enantiomer is available only via the hydrolytic approach.
The other enzymatic routes
5,21 to (+)-
1 follow roughly similar procedures. Both employ pH 7 buffer solutions of the hydrolytic enzyme porcine pancreas lipase (PPL) as opposed to electric eel
acetyl cholinesterase (EEAC) used here. Although EEAC does afford better enantioselection than PPL (96% vs. 92% e.e.
21), this apparent advantage may be viewed as somewhat tenuous since (+)-
1 can be optically enriched via repeated recrystallizations. However, EEAC is clearly superior to PPL in terms of chemical yield. PPL mediated hydrolyses lead to yields of (+)-
1 in the
50% range (plus recovered starting material), while the EEAC conversion rate is
90–95%. More importantly, the EEAC route eliminates the need to recycle starting material.
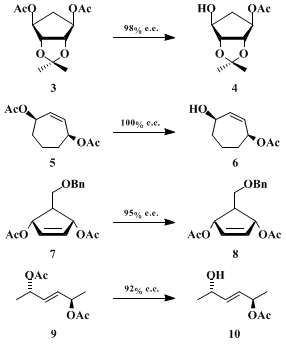
EEAC has been used successfully in other enantiotopic differentiations. Johnson, et al.
22 have reported that diester
3 can be readily transformed into hydroxy acetate
4 via this enzymatic process in 98% e.e. and an 80% chemical yield. Similarly, hydroxy acetate
6 was prepared from its parent diester
5 by Pearson, et al.
23 in 100% e.e., although in 39% yield (50–55% recovered starting material). The enzyme also appears effective on 4-substituted cis-3,5-diacetoxycyclopentenes as Danishefsky
24 demonstrated with the conversion of
7 into
8 in 95% yield and 95% e.e. Finally, the successful enantioselective hydrolysis of
9 into
10 (77%, 92% e.e.) extends the range of useful EEAC substrates to acyclic cases.
25Immobilized EEAC (Sigma Chemical Company) has been found equally effective and allows economical, large-scale hydrolyses of 2. The immobilized enzyme, which is covalently bonded to agarose beads, is easily recovered through centrifugation. Reuse does not compromise the enantioselectivity (unpublished results).
This preparation is referenced from:
Appendix
Chemical Abstracts Nomenclature (Collective Index Number);
(Registry Number)
brine
acetyl cholinesterase
EEAC
hydrochloric acid (7647-01-0)
ethyl acetate (141-78-6)
ether,
ethyl ether (60-29-7)
acetic anhydride (108-24-7)
sodium hydroxide (1310-73-2)
chloroform (67-66-3)
sodium bicarbonate (144-55-8)
sucrose
Pentane (109-66-0)
sodium azide (26628-22-8)
methylene chloride (75-09-2)
magnesium sulfate (7487-88-9)
Imidazole (288-32-4)
argon (7440-37-1)
sodium dihydrogen phosphate (7558-80-7)
calcium hydride (7789-78-8)
p-anisaldehyde (123-11-5)
4-oxo-2-cyclopentenyl acetate (768-48-9)
phosphorus pentoxide (1314-56-3)
(1R,4S)-(+)-4-Hydroxy-2-cyclopentenyl acetate,
4-Cyclopentene-1,3-diol, monoacetate, (1R-cis)-,
(±)-cis-4-Hydroxy-2-cyclopentenyl acetate (60410-16-4)
cis-3,5-Diacetoxycyclopentene (54664-61-8)
(±)-cis-4-acetoxy-1-hydroxycyclopent-2-ene (60410-18-6)
sodium dihydrogen phosphate monohydrate (10049-21-5)
cis-3,5-Dihydroxycyclopentene
(R)-(+)-α-methoxy-α-(trifluoromethyl)phenylacetic acid (20445-31-2)
Copyright © 1921-, Organic Syntheses, Inc. All Rights Reserved