Org. Synth. 1992, 70, 35
DOI: 10.15227/orgsyn.070.0035
(R)-(−)-10-METHYL-1(9)-OCTAL-2-ONE
[2(3H)-Naphthalenone, 4,4a,5,6,7,8-hexahydro-4a-methyl-, (R)-]
Submitted by G. Revial and M. Pfau
1.
Checked by Graham N. Maw and Robert K. Boeckman, Jr..
1. Procedure
A. Imines (1). In a 1000-mL, round-bottomed flask, fitted with a toluene-filled Dean-Stark water separator, 100.0 g (0.825 mol) of (S)-(−)-α-methylbenzylamine (Note 1), 92.5 g (0.825 mol) of 2-methylcyclohexanone (Note 2) and 100 mL of toluene are heated at reflux temperature under a nitrogen atmosphere. After 24 hr, ca. 15 mL of water (ca. 100% theor.) has been removed azeotropically.
B. (R)-(+)-2-Methyl-2-(3-oxobutyl)cyclohexanone (2). The solution of imines 1 is cooled in an ice bath and 72.5 mL (61.0 g, 0.870 mol, ca. 1.05 equiv) of freshly distilled methyl vinyl ketone (Note 3) is added with a syringe, with magnetic stirring, under a nitrogen atmosphere. The flask is then heated at ca. 40°C for 24 hr.
The slightly yellow solution is cooled in an ice bath and 60 mL of glacial acetic acid (ca. 1 mol) and 50 mL of water are added. Hydrolysis is achieved by stirring the heterogeneous mixture at room temperature for 2 hr. The now clear solution is poured into a 2000-mL separatory funnel containing 100 mL of brine and 160 mL of water and is extracted five times with a 50:50 mixture of ether-petroleum ether (35–60°C) (1000-mL total amount). The organic phase is washed efficiently with 20 mL of 10% hydrochloric acid, 20 mL of water, and two 10-mL portions of brine.
The aqueous layer is kept for recovery of the amine (see D below).
The pale yellow organic layer is dried over a small amount of anhydrous magnesium sulfate and filtered. The solvents are removed with a rotary evaporator at ca. 40°C and the crude diketone 2 (ca. 145 g) is used directly for the next step.
C.
(R)-(−)-10-Methyl-1(9)-octal-2-one (3). Dry
methanol (600 mL) is added to the crude diketone
2 contained in a
2000-mL, round-bottomed flask and a
rubber septum is fitted. The solution is stirred at room temperature for 15 min under a slight stream of
nitrogen to remove traces of
oxygen. A
25 wt. % solution of sodium methoxide in methanol (Note 4) is then introduced dropwise with a syringe, under stirring, until a slightly red color develops
(Note 5). At this point,
15 mL (ca. 0.07 mol) of the sodium methoxide solution is added and the mixture is heated at 60°C for 10 hr under a
nitrogen atmosphere. The solution is cooled and the now deep red solution is neutralized with
glacial acetic acid (ca. 4.5 mL) until the color turns yellow.
Methanol is removed with a rotary evaporator until a thick paste (
sodium acetate) results; this is dissolved with 200 mL of water. Extraction is effected in a 2000-mL separatory funnel using four portions of a
50:50 mixture (1000 mL total amount) of ether-petroleum ether (35–60°C). The pale orange organic phase is washed twice with 40 mL of water, then twice with
20 mL of brine. The organic layer is dried over a small amount of anhydrous
magnesium sulfate and filtered. The solvents are removed with a rotary evaporator at ca. 40°C and the red oily residue of crude octalone
3 is distilled under reduced pressure to afford ca.
110 g of a colorless oil, bp
70°C (2 mm) (Note 6). The oil is purified by recrystallization at a low temperature with an apparatus designed for this purpose.
2 The oil is dissolved with stirring in a 1000-mL, round-bottomed flask with
370 mL of pentane under a
nitrogen atmosphere. The flask is then cautiously cooled in a Dewar flask containing liquid
nitrogen until the solution is solid. The Dewar flask is removed and the temperature is allowed to rise, while magnetic stirring is resumed. When a crystalline mass in suspension appears suddenly
(Note 7), an
acetone bath adjusted to about −10°C is installed. When only a few crystals remain in suspension, the bath temperature is slowly lowered to −35°C, inducing crystallization. After the mixture has been kept at −35°C for 15 min, magnetic stirring is stopped and efficient filtration is performed through the filter stick. Still at −35°C, the compound is washed with
40 mL of pentane. The solution is stirred vigorously for 5 min and filtered as before. The washing operation is repeated two more times. The flask is warmed to room temperature and the oil is distilled as before, bp
70°C (2 mm), to afford
60–65 g (
44–48% overall yield from
2-methylcyclohexanone) of methyloctalone
3 [>99% chemical purity, capillary GLC
(Note 6)],
[α]D20 −210° (
ethanol,
c 1.00), optical purity 96%
(Note 8).
D. Recovery of (S)-(−)-α-methylbenzylamine. The aqueous layer (see above, B, 3rd paragraph) contained in a 1000-mL flask is cooled in an ice bath under a nitrogen atmosphere. An aqueous 10% sodium hydroxide solution is added with stirring until a pH of 12–14 (Note 9) is reached. The mixture is transferred to a 2000-mL separatory funnel and extracted three times with ether (1000-mL total amount). The organic phase is washed twice with 100 mL of water and twice with 30 mL of brine, then dried over potassium carbonate and filtered. The solution is concentrated at room temperature with a rotary evaporator and distilled under reduced pressure to afford 85–90 g (85–90% yield) of the amine, bp 70°C (15 mm), the specific rotation of which is the same as that of the original starting material.
2. Notes
1.
(S)-(−)-α-Methylbenzylamine (95.8% optical purity) and its (R)-(+)-enantiomer (93.4% optical purity) were purchased from Aldrich Chemical Company, Inc. and used without further purification.
CAUTION: These amines are toxic. Air must be excluded to prevent formation of carbonates.
2.
2-Methylcyclohexanone was purchased from Aldrich Chemical Company, Inc., and used without further purification.
3.
Methyl vinyl ketone, purchased from Aldrich Chemical Company, Inc., is dried over anhydrous
potassium carbonate for 0.5 hr. The now slightly-colored oil is filtered and distilled under a
nitrogen atmosphere at reduced pressure, bp
55°C (260 mm).
The colorless center of the distillate is collected over a few milligrams (ca. 0.05% concentration) of hydroquinone.
4.
A
25 wt % solution of sodium methoxide in methanol (4.6 M) was purchased from Aldrich Chemical Company, Inc.5.
Any
acetic acid present is thus neutralized (a few drops are usually sufficient). Confirm that a basic pH has been reached by testing a drop of the solution, removed by syringe.
6.
Although a TLC test (
silica gel Merck 60 F
254,
ethyl acetate/hexane 20:80 elution, UV and chromic-sulfuric acid visualization) of this oil reveals only one spot (R
f 0.6), capillary GLC (Hewlett-Packard HP-5, 25 m × 0.2 mm × 0.5 μm, 200°C) shows in addition to methyloctalone
3 (retention time, 10.9 min), an 8% impurity (retention time, 11.7 min) which is probably the regioisomer
4.
A chemically pure sample of methyloctalone 3, [α]D20 −190° (ethanol, c 1.00) is obtained by preparative GLC (3 m × ¼", 5% carbowax 20 M on Aeropak 30, 180°C, retention time 12.4 min) of a distillate sample. The impurity has a 13.2-min retention time under these conditions. A good approximation of the reaction ee is calculated as follows, taking into account the optical purity of the starting amine (95.8%) and the [α]D20 −219° (ethanol, c 1.00) value (Note 8) for optically pure methyloctalone 3: ee = 100 × 190 × 100/95.8 × 219 ≅ 91%.
7.
If no crystalline mass is formed but instead an emulsion of the two liquid phases appears, the entire solidification must be repeated (liquid
nitrogen cooling).
8.
Chiral capillary GLC (OV-1701 + trimeb/2), 25 m × 0.3 mm, He, 65 kPa, 130°C: (R)-(−)-
3, 58.3 min (98%); (S)-(+)-
3, 62.0 min (2%). Optically pure methyloctalone
3 is obtained by recrystallizing a sample of the purified compound several times until a constant
[α]D20 −219° (
ethanol,
c 1.00 or
methanol,
c 1.10) is observed. As chiral capillary GLC shows only (R)-(−)-
3, the
[α]D20 −219° (
methanol,
c 1.1) value from the literature
3 is confirmed.
9.
If a pH lower than 12 is used, recovery of the amine is poor.
Handling and Disposal of Hazardous Chemicals
The procedures in this article are intended for use only by persons with prior training in experimental organic chemistry. All hazardous materials should be handled using the standard procedures for work with chemicals described in references such as "Prudent Practices in the Laboratory" (The National Academies Press, Washington, D.C., 2011 www.nap.edu). All chemical waste should be disposed of in accordance with local regulations. For general guidelines for the management of chemical waste, see Chapter 8 of Prudent Practices.
These procedures must be conducted at one's own risk. Organic Syntheses, Inc., its Editors, and its Board of Directors do not warrant or guarantee the safety of individuals using these procedures and hereby disclaim any liability for any injuries or damages claimed to have resulted from or related in any way to the procedures herein.
3. Discussion
Optically active methyloctalone
3 (or its enantiomer) has been obtained by resolution
3,4 and by asymmetric synthesis.
5 Both enantiomers are also available commercially. The synthesis described here is by far the most simple and gives the best yield. It is an application of the general method reported for the enantioselective elaboration of quaternary carbon centers through Michael-type alkylation of chiral imines.
6 7 8 9 10 11 12 13 14This general method has the following advantages: very simple procedures, very mild conditions involving moderate reaction temperatures and no acids, bases, catalysts, etc., allowing the use of labile reactants, excellent regio- and enantioselectivities as well as high chemical yields, and creation of useful chiral building blocks with quaternary carbon centers involving functionalized chains, allowing one to devise syntheses of natural compounds of various types (terpenes, steroids, alkaloids, etc.). As both optically active amines are commercially available, targets with either desired absolute configuration can be synthesized. Easy recovery of the auxiliary chiral moiety is possible.
Racemic methyloctalone
3 has been used as a starting material for many transformations as well as for syntheses of racemic natural compounds. In the following examples, the absolute configuration of the methyloctalone that would be required to obtain the natural enantiomer is indicated in parentheses:
β-gorgonene15 (S),
widdrol16,17 (R),
thujopsene16,18 (R),
7-hydroxycostal19,20 (R),
β-selinene21,22 (R),
costol,
costal and
costic acid21 (R),
norketoagarofuran23,24 (R),
β-eudesmol21,22,23,25 (R), taxane model
26 (S),
polygodial27 (S),
warburganal27 (S).
(R)-Methyloctalone
3 itself has been used to synthesize a pasmalinine model,
28 a subunit of
penitrem D,
29 and several biologically active decalin derivatives.
30The following key-intermediates have been prepared by a procedure similar to the one described above, generally with excellent chemical and optical yields :
The use of substituted acrylic acid derivatives shows that besides the high enantioselectivity usually observed, the process is also highly diastereoselective, leading to compounds such as 27-32.
An acetylenic electrophile such as methyl propiolate leads to unsaturated compounds 35 and 36.
Linear β-ketoesters and lactones lead to compounds such as 37-39.
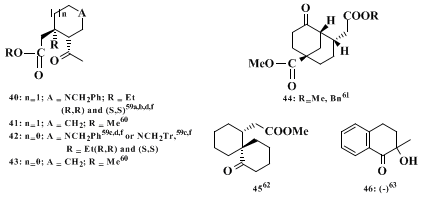
Compounds 40-45 arise from an analogous but intramolecular reaction. A special application of the chiral imine procedure, i.e., the use of an oxaziridine rather than an electrophilic olefin, yielding compound 46, has been reported.
Several of these chiral non racemic compounds were used for the synthesis of bilogically active products. Thus, (R)- and (S)-dimethyloctalones
5 lead respectively to natural and ent-geosmin.
31 The (R)-enantiomer is also a precursor for the formal synthesis of
isocostic and 3-isocostic acid32 while the (S)-enantiomer would lead to
tuberiferine33 and was actually used to synthesize
(−)-TMD, a specific inhibitor of the cholesterol biosynthesis.
34 (S,S)-Dimethyloctalone
6 (a natural compound) leads to a natural
octalin31 and would lead to
α-vetispirene35 while the (R,R)-enantiomer would lead to
frullanolide.
36 Compound
7 (R,R) is a precursor for the formal synthesis of
muzigadial27 while the (S,S)-enantiomer would lead to
polygodial27 and
warburganal.
27 (+)-α-Cyperone 8 has been widely used as a starting material for the synthesis of many other sesquiterpenes
37 and was in particular converted to
ent-allohedycaryol.
38 Compound
9 was transformed into a C
2-symmetric chiral dione
39 while compound
10 was the starting material for the synthesis of (−)-polywood.
34 Tricyclic compounds
11 and
12 were used respectively to synthesize a ring C aromatic steroid
40 and a key [ABC] steroid intermediate
41 while compounds
13 were transformed into isosteres of aphidicolin as potential antiherpes agents.
42 Methoxyketone
14 was converted to a precursor of
bruceantin43 and compound
15 was used for the preparation of a 14-hydroxymorphinan derivative.
44 (R)-Ketoester
16 is a key building block for the synthesis of
(+)-cassiol,
45 a potent antiulcerogenic agent,
tridensone,
46 and
(−)-19-noraspidospermidine,
47 while
17 led to
(+)-aspidospermidine.
48 Compound
18 was converted to
(−)-nitramine.
49,14 Lactams
19 show potent inhibition of the recombinant human type-1 steroid
5α-reductase. Compound
20 was used to perform the first synthesis of
(+)-petasine and
(+)-isopetasine, potent antiinflammatory and spasmolytic agents.
50 Ketoester
21 is an intermediate for the formal total synthesis of
vallesamidine.
51 Ketodiesters
22 or
23 led to the marine antibiotic
(−)-malyngolide.
52 Aza-heterocycles
24 were used to synthesize substituted isoquinoline derivatives which were evaluated as potential inhibitors of
2,3-oxidosqualene lanosterol cyclase53 while oxo-heterocycles
25 and
26 have been used respectively for the synthesis of the
mycotoxin,
(−)vertinolide54 and of a subunit of marine polyethers toxins
55 as well as to advanced intermediates for the synthesis of the anti-fertility agent,
zoapatanol.
56 Compounds
32 and
34 lead to precursors of
Vinca and
Hunteria alkaloids.
57 Ethylenic compounds
35 and
36 were used respectively for the total synthesis of both riccardiphenols A and B
58 and
isocuparene.
59 Acyclic compound
39 was converted to a nipecotic acid derivative.
60 (R,R)-
40 is a common intermediate in the total synthesis of
(−)-ajmalicine and
(−)-tetrahydroalstonine61,62,63 as well as of
(+)-yohimbine,
64,62,63 (−)-ochropposinine,
63 and
(−)-emetine62,63 while (S,S)-
40 leads to
(−)-(10R)-hydroxydihydroquinine.
61,62,63
Appendix
Chemical Abstracts Nomenclature (Collective Index Number);
(Registry Number)
silica gel
petroleum ether
brine
(S)-(−)-α-methylbenzylamine
β-gorgonene
widdrol
thujopsene
7-hydroxycostal
β-selinene
costol
costal
costic acid
norketoagarofuran
β-eudesmol
polygodial
warburganal
penitrem D
isocostic and 3-isocostic acid
tuberiferine
(−)-TMD
octalin
α-vetispirene
frullanolide
muzigadial
(+)-α-Cyperone
ent-allohedycaryol
bruceantin
(+)-cassiol
tridensone
(−)-19-noraspidospermidine
(+)-aspidospermidine
(−)-nitramine
5α-reductase
(+)-petasine
(+)-isopetasine
vallesamidine
(−)-malyngolide
2,3-oxidosqualene lanosterol cyclase
mycotoxin
(−)vertinolide
zoapatanol
isocuparene
(−)-ajmalicine
(−)-tetrahydroalstonine
(−)-ochropposinine
(−)-emetine
(−)-(10R)-hydroxydihydroquinine
ethanol (64-17-5)
potassium carbonate (584-08-7)
hydrochloric acid (7647-01-0)
acetic acid (64-19-7)
ethyl acetate (141-78-6)
methanol (67-56-1)
ether (60-29-7)
sodium acetate (127-09-3)
sodium hydroxide (1310-73-2)
hydroquinone (123-31-9)
oxygen (7782-44-7)
nitrogen (7727-37-9)
sodium methoxide (124-41-4)
toluene (108-88-3)
Pentane (109-66-0)
magnesium sulfate (7487-88-9)
hexane (110-54-3)
methyl vinyl ketone (78-94-4)
2-methylcyclohexanone (583-60-8)
oxaziridine
methyl propiolate (922-67-8)
(R)-(−)-10-METHYL-1(9)-OCTAL-2-ONE,
2(3H)-Naphthalenone, 4,4a,5,6,7,8-hexahydro-4a-methyl-, (R)- (63975-59-7)
(R)-(+)-2-Methyl-2-(3-oxobutyl)cyclohexanone (91306-30-8)
(+)-yohimbine
Copyright © 1921-, Organic Syntheses, Inc. All Rights Reserved