Org. Synth. 1992, 70, 256
DOI: 10.15227/orgsyn.070.0256
SYNTHESIS OF β-KETO ESTERS BY C-ACYLATION OF PREFORMED ENOLATES WITH METHYL CYANOFORMATE: PREPARATION OF METHYL (1α,4aβ,8aα)-2-OXODECAHYDRO-1-NAPHTHOATE
Submitted by Simon R. Crabtree, Lewis N. Mander, and S. Paul Sethi
1.
Checked by Thierry Vandenheste and Leo A. Paquette.
1. Procedure
CAUTION! Cyanide salts are formed in this procedure. All procedures should be conducted in a well-ventilated hood and rubber gloves should be worn.
Liquid ammonia (350 mL) is dried over sodium amide for 20 min (Note 1), then distilled under a positive pressure of dry nitrogen into a flame-dried, 1-L, three-necked, round-bottomed flask equipped with a dry ice condenser, rubber septum and magnetic stirring bar. The flask is cooled to −78°C and small pieces of lithium (1.11 g, 0.16 mol) (Note 2) are added over 10 min to afford a deep blue solution. A solution of enone 1 (10.08 g, 0.07 mol) (Note 3) in tert-butyl alcohol (5.10 g, 0.07 mol) (Note 4) and ether (40 mL) (Note 5) is added dropwise over a 15-min period at −78°C, and then sufficient isoprene (Note 6) is added dropwise to discharge the residual blue color of the reaction mixture. The ammonia is allowed to evaporate under a stream of dry nitrogen and the ether is removed under reduced pressure to leave a white foam. After a further 5 min under high vacuum, the nitrogen atmosphere is restored, the lithium enolate 2 is suspended in dry ether (80 mL) at −78°C by vigorous stirring, and methyl cyanoformate (6.73 g, 0.08 mol) (Note 7) is added over a 5-min period. Stirring is maintained at this temperature for a further 40 min and then the mixture is allowed to warm to 0°C. Water (500 mL) and ether (200 mL) are added and the mixture is stirred vigorously until the precipitate has dissolved. After separation of the organic layer, the aqueous phase is extracted with ether (2 × 300 mL), the combined extracts are washed with water (200 mL) and brine (200 mL) (Note 8), and then dried over anhydrous magnesium sulfate. Removal of solvent under reduced pressure furnishes a pale yellow oil which is dissolved in hexane (200 mL); the solution is kept in the freezer (−28°C) for approximately 1.5 hr, after which time very pale yellow crystals of the β-keto ester 3 are deposited. These are collected by filtration at low temperature and after concentration of the mother liquors, two further crops are collected. Flash chromatography (ethyl acetate/hexane, 13:87) on silica gel (140 g) (Note 9) of the residue (3.5 g) from the remaining mother liquor followed by two recrystallizations affords further 3. The total quantity of 3 obtained is 11.5–11.8 g (81–84% yield) (Note 10).
2. Notes
1.
Sodium amide is generated in situ from
sodium metal and a crystal of
ferric nitrate.
Sodium metal by itself is not an effective drying agent.
2.
Lithium wire was purchased from Merck Schuchardt.
4.
tert-Butyl alcohol was purchased from Ajax Chemicals and was freshly distilled from
calcium hydride.
5.
Diethyl ether was purchased from Ajax Chemicals and was freshly distilled from
sodium and
benzophenone.
6.
Isoprene was purchased from Eastman Kodak, Ltd.
7.
Methyl cyanoformate was purchased from the Aldrich Chemical Company, Inc. The ethyl and benzyl analogues are also available. Small quantities up to 30 g are conveniently prepared by the method of Childs and Weber,
2 but several workers have found it to be unsatisfactory on a larger scale because of the generation of inseparable impurities.
8.
The aqueous washings contain toxic
lithium cyanide and should be treated with a strong oxidizing agent (e.g.,
potassium permanganate) before disposal.
9.
Merck Kieselgel 60
silica gel was used.
10.
A sample recrystallized twice from
hexane had mp
48–49°C (Anal. Calcd for C
12H
18O
3: C, 68.53; H. 8.63%; Found C, 68.59; H, 8.93). The spectroscopic properties of this product are as follows: IR (CHCl
3) cm
−1: 3026, 2931, 2859, 1744, 1710, 1454, 1447, 1437, 1364, 1348, 1340, 1155, 1112;
1H NMR (CDCl
3) δ: 1.00–1.50 (m, 6 H), 1.65–1.8 (m, 5 H), 1.9–2.0 (m, 1 H CH), 2.30–2.50 (m, 2 H, CH
2), 3.12 (d, 1 H, J = 12), 3.76 (s, 3 H, OMe).
13C NMR (CDCl
3) δ: 25.4, 25.9, 32.6, 32.9, 33.3, 40.5, 41.4, 45.5, 51.8, 53.8, 170.2, 205.6; MS (m/z): 210 (M
+, 34.6%), 192 (72), 122 (66), 108 (59), 95 (68), 94 (94), 81 (100), 79 (53), 67 (77), 51 (61).
Handling and Disposal of Hazardous Chemicals
The procedures in this article are intended for use only by persons with prior training in experimental organic chemistry. All hazardous materials should be handled using the standard procedures for work with chemicals described in references such as "Prudent Practices in the Laboratory" (The National Academies Press, Washington, D.C., 2011 www.nap.edu). All chemical waste should be disposed of in accordance with local regulations. For general guidelines for the management of chemical waste, see Chapter 8 of Prudent Practices.
These procedures must be conducted at one's own risk. Organic Syntheses, Inc., its Editors, and its Board of Directors do not warrant or guarantee the safety of individuals using these procedures and hereby disclaim any liability for any injuries or damages claimed to have resulted from or related in any way to the procedures herein.
3. Discussion
The present procedure provides an example of the regiospecific synthesis of β-keto esters by the C-acylation of preformed lithium enolates with cyanoformate esters.
3 The enolates can be generated in a variety of ways, including direct enolization of ketones with suitable bases, liberation from silyl enol ethers and acetates, conjugate additions of cuprates to α,β-unsaturated enones and by the reduction of enones by
lithium in liquid
ammonia, as in the example described above. The more traditional acylating agents (acyl halides and anhydrides) afford variable amounts of O-acylated products
4 and although the use of reaction temperatures of −100°C is reported to obviate this problem with some substrates,
5 the method does not appear to be general. The classical solution has been to use
carbon dioxide followed by esterification,
6 but yields rarely exceed 50%, indicating,
inter alia, that the formation of enol carbonates probably occurs to a significant extent and that these unstable intermediates decompose to ketones on work up.
7 There is also the problem of handling thermally unstable β-keto acids.
Carbon disulfide8 and
carbon oxysulfide9 have been used instead of
carbon dioxide, and the products methylated in situ to form dithio and thio esters, respectively. These can subsequently be converted into methyl esters by
mercury(II) diacetate-catalyzed transesterification in
methanol, but the methods are elaborate and do not appear to have found general use. Diethyl dicarbonate and its analogues show promise as possible alternatives to cyanoformates, but the reported yields to date are inferior.
10In the early investigations of this reaction it was found that in one comparative study of lithium, sodium and potassium enolates, only the lithium derivatives reacted satisfactorily,
3 although sodium enolates have subsequently been used successfully.
11 A range of cyanoformates can be employed, but the tert-butyl derivatives have been reported to be too unreactive. However, a satisfactory substitute which affords esters that can be de-alkylated with comparable facility is the p-methoxybenzyl analogue,
12 13 although it is prepared with some difficulty. In general, benzyl derivatives seem to give slightly better yields, possibly due to the more efficient isolation of products from substrates with lower molecular weights. Optically active cyanoformates derived from
(+)-menthol,
(−)-borneol, and the Oppolzer alcohol were reported to furnish good chemical yields, but the level of stereoselectivity was disappointingly low.
14For more conformationally-constrained chiral substrates, however, diastereoselectivity can be expected to be good to excellent. Lithium enolates derived from sterically unencumbered cyclohexanones undergo preferential axial acylation as illustrated by the reductive acylation of
(R)-(−)-carvone 4 to afford a 3:1 mixture of esters
5 and
6, whereas equatorial acylation is favored in compounds that possess an alkyl substituent in a 1,3-syn-axial relationship to the reacting center, as in the conversion of tricyclic enone
7 to ester
8 (epimeric with the product from the more traditional sequence of acylation followed by alkylation).
15 (In substrates of this kind it is assumed that the transition state structure is based on a twist-boat conformation which permits the reagent to approach along an axial-like trajectory on the less encumbered, lower face of the substrate.)
16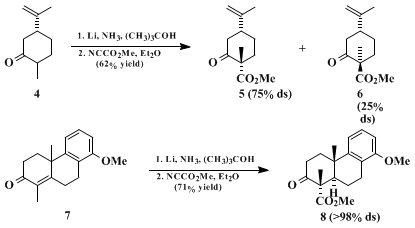
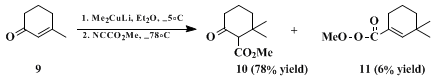
For compounds in which the β-carbon of the enolate is sterically hindered, this problem can normally be eliminated by the use of diethyl ether as the solvent. In several cases a switch from exclusive O-acylation in tetrahydrofuran to complete C-acylation in ether has been observed, and the conversion of 7 into 8 provides a typical illustration. When 3-methyl-2-cyclohexen-1-one 9 is converted into 10 by the addition of lithium dimethyl cuprate followed by in situ reaction with methyl cyanoformate, however, enol carbonate 11 accounts for 6% of the products. This can be avoided if the intermediate enolate is trapped as the enol trimethylsilyl ether and the enolate reliberated by treatment with butyllithium (note that the traditional reagent, methyllithium, is ineffective in ether as a solvent), but the overall yield is reduced.
Quite apart from the issue of regioselectivity, the present method is exceptionally reliable and makes it possible to prepare β-keto esters from ketones under especially mild conditions. It is not only the method of choice with sensitive substrates, but will often ensure superior results with more stable intermediates as well.
8,17 A wide range of examples is provided in the Table, while further examples have been reported elsewhere.
18 19 20 The methodology has also been applied to esters,
12,13 lactones,
21 phosphonates,
22 23 imines
24 and the N-acylation of lactams.
25 The formation of β-diketones by the treatment of lithium enolates with alkanoyl and aroyl nitriles has also been described.
26
TABLE
PREPARATION OF β-KETO-ESTERS BY THE KINETICALLY CONTROLLED C-ACYLATION OF LITHIUM ENOLATES WITH METHYL CYANOFORMATE
|
|
Entry
|
Substrate
|
Procedure‡
|
Product
|
Yield(%)
|
|
|
1
|
|
A
|
|
84
|
86
|
|
2
|
|
A
|
|
86
|
86
|
|
3
|
|
A
|
|
96
|
|
4
|
|
A
|
|
85
|
|
5
|
|
A
|
|
71
|
75
|
|
6
|
|
B
|
|
72
|
68
|
|
7
|
|
A
|
|
65
|
|
8
|
|
A
|
|
84
|
|
9
|
|
A
|
|
92
|
|
10
|
|
A
|
|
92
|
|
11
|
|
C
|
|
85
|
|
‡Procedures: A: Reference; B: Lithium enolates generated by addition of methyllithium in THF at −78°C; C: As described for the conversation of enone 1 into β-keto ester 3.
|
Appendix
Chemical Abstracts Nomenclature (Collective Index Number);
(Registry Number)
silica gel
carbon oxysulfide
brine
(R)-(−)-carvone
(−)-borneol
lithium dimethyl cuprate
ammonia (7664-41-7)
ethyl acetate (141-78-6)
methanol (67-56-1)
ether,
diethyl ether (60-29-7)
potassium permanganate (7722-64-7)
nitrogen (7727-37-9)
mercury(II) diacetate (1600-27-7)
carbon dioxide (124-38-9)
Benzophenone (119-61-9)
sodium (13966-32-0)
carbon disulfide (75-15-0)
(+)-menthol (15356-60-2)
lithium (7439-93-2)
magnesium sulfate (7487-88-9)
sodium amide (7782-92-5)
butyllithium (109-72-8)
Tetrahydrofuran,
THF (109-99-9)
ferric nitrate
ISOPRENE (78-79-5)
hexane (110-54-3)
Methyllithium (917-54-4)
tert-butyl alcohol (75-65-0)
calcium hydride (7789-78-8)
3-methyl-2-cyclohexen-1-one
lithium cyanide
METHYL CYANOFORMATE (17640-15-2)
Methyl (1α,4aβ,8aα)-2-oxodecahydro-1-naphthoate (147086-16-6)
Copyright © 1921-, Organic Syntheses, Inc. All Rights Reserved