Checked by Gregory L. Aaron, Matthew M. Davis, and Kay M. Brummond.
1. Procedure
2. Notes
1.
L(-)-Proline (99+%) was used as received from Acros Organics.
2.
Chloroform (ACS grade) was used without further purification from Fisher Scientific.
3.
The original procedure reported by Germanas employed trichloroacetaldehyde or chloral. However, this reagent is regulated and difficult to obtain. The submitters have found that commercially available 2,2,2-trichloro-1-ethoxyethanol can be used as a masked form of chloral.
4.
2,2,2-Trichloro-1-ethoxyethanol (98%) was used as commercially available and was obtained from Alfa Aesar.
5.
Disappearance of L(-)-Proline (R
f = 0.89) and formation of (3
R,7a
S)-3-(trichloromethyl)tetrahydropyrrolo[1,2-c]oxazol-1(3
H)-one (R
f = 0.26) was observed via reverse phase thin layer chromatography performed on Partisil
® KC18 Silica Gel 60 (200 μm thickness) on glass backed plates (1:1 H
2O/CH
3CN) visualizing with KMnO
4 TLC Stain (yellow spots). The reaction requires 15-19 h to reach completion, during which time a color change from a milky opaque to an orange solution is observed.
6.
(3
R,7
aS)-3-(Trichloromethyl)tetrahydropyrrolo[1,2-
c]oxazol-1(3
H)-one
2,3 displays the following physical and spectral characteristics: mp 108-109 °C (lit.
3 107-109 °C); optical rotation: [α]
D = +34.0 (
c 2, C
6H
6), lit.
3 [α]
D = +33 (
c 2, C
6H
6);
1H NMR
pdf (500 MHz, CDCl
3) δ: 1.70-1.79 (m, 1 H), 1.90-1.97 (m, 1 H), 2.08-2.14 (m, 1 H), 2.19-2.27 (m, 1 H), 3.11-3.15 (m, 1 H), 3.42 (ddd,
J = 11, 7.5, 6 Hz, 1 H), 4.12 (dd,
J = 9, 4.5 Hz, 1 H), 5.17 (s, 1 H);
13C NMR
pdf (100 MHz, CDCl
3) δ: 25.3, 29.9, 57.9, 62.4, 100.6, 103.6, 175.5; IR (thin film) 2978, 2962, 2899, 2871, 1782, 1327, 1178, 1009, 959, 815, 791, 744 cm
-1; Anal. Calcd for C
7H
8Cl
3NO
2: C, 34.39; H, 3.30; N, 5.73. Found: C, 34.47; H, 3.28; N, 5.65.
7.
Unlike the Seebach pivaldehyde/proline condensate, this product is air- and moisture-stable and can be stored upon the bench top with no decomposition by NMR spectroscopy after more than 30 days.
8.
Following the submission of this procedure,
(3R,7aS)-3-(trichloromethyl)tetrahydropyrrolo[1,2-
c]oxazol-1(3
H)-one is now commercially available.
9.
N,N-Diisopropylamine (99%) was purchased from Fisher Scientific and was freshly distilled from CaCl
2 prior to use.
10.
Tetrahydrofuran (THF, 99.5%) was purchased from Sigma-Aldrich and was purified via a Sol-Tek ST-002 solvent purification system.
11.
1.6 M
n-Butyllithium in hexanes was purchased from Sigma-Aldrich and freshly titrated using the method developed by Love and Jones.
412.
A color change is apparent as the enolate is formed. The LDA solution changes from light yellow, to dark red, to dark brown upon the addition of the oxazolinone.
13.
Allyl bromide (98%) was used as received from Alfa Aesar.
14.
Thin layer chromatography (TLC) on silica gel F
254 (200 μm thickness) glass backed plates was used to monitor the alkylation. Developing the plate in 1:7 EtOAc:hexanes separates the product (R
f = 0.44) from the starting material (R
f = 0.27). Both the product and starting material can be visualized with KMnO
4 TLC stain (yellow spots).
15.
The allylated product is of sufficient purity to be used in the next step. However, an analytical sample was obtained by purifying 145 mg of the crude material via flash silica gel chromatography (Column inner diameter 1 cm; packed length 12.5 cm) eluting 1:7 EtOAc/hexanes to afford 111 mg of alkylated product. (3
R,7
aR)-7
a-Allyl-3-(trichloromethyl)-tetrahydropyrrolo[1,2-
c]oxazol-1(3
H)-one displays the following physical and spectral characteristics: pale yellow oil, which slowly crystallizes upon standing at -10 °C; optical rotation: [α]
D = +44 (
c 2, CHCl
3), [α]
D = +60 (
c 2, C
6H
6), lit.
3 [α]
D = +44.6 (
c 2, CHCl
3);
1H NMR
pdf (500 MHz, CDCl
3) δ: 1.60-1.70 (m, 1 H), 1.86-1.93 (m, 1 H), 2.02 (ddd,
J = 13.5, 10.0, 7.0 Hz, 1 H), 2.13 (ddd,
J = 13.0, 8.0, 3.0 Hz, 1 H), 2.55 (dd,
J = 14.0, 8.5 Hz, 1 H), 2.62 (dd,
J = 14.0, 6.5 Hz, 1 H), 3.14-3.24 (m, 2 H), 4.98 (s, 1H), 5.17 (d,
J = 6.0 Hz, 1 H), 5.20 (s, 1 H), 5.85-5.93 (m, 1 H);
13C NMR
pdf (100 MHz, CDCl
3) δ: 25.2, 35.2, 41.5, 58.3, 71.3, 100.4, 102.3, 119.9, 131.9, 176.2; IR (thin film) 3079, 2976, 2897, 1801, 1640, 1457, 1438, 1355, 1324, 1192, 1129, 1104, 1020, 922, 838, 803, 746 cm
-1; Anal. Calcd for C
10H
12Cl
3NO
2: C, 42.21; H, 4.25; N, 4.92. Found: C, 42.51; H, 4.31; N, 4.84.
16.
Methanol (MeOH, ACS grade) was used as received and obtained from Fischer Scientific. Anhydrous MeOH can be employed, but the yield was unchanged as observed by the submitters.
17.
Sodium metal cubes (99.95%) in mineral oil were supplied by Sigma-Aldrich. The sodium metal was cut into small pieces using a razor blade and weighed into a tared beaker containing hexanes to remove residual mineral oil prior to addition.
18.
The deprotection was monitored by TLC using silica gel F
254 (200 μm thickness) glass backed plates, 1:5 EtOAc:hexanes, KMnO
4 TLC stain (yellow spots) observing consumption of the starting material (R
f = 0.57) and formation of the
N-formyl ester intermediate (R
f = 0.11). It was observed that a full equivalent of sodium methoxide is not necessary for the opening of the lactone to the
N-formyl ester intermediate.
19.
Acetyl chloride (98%) was used as received from Sigma-Aldrich.
20.
The addition of acetyl chloride must be conducted at a slow rate to avoid an exothermic reaction and loss of HCl gas. The submitters observed that if the addition is too fast, an additional quantity of
acetyl chloride (~20 mL) generally has to be added to the reaction mixture once the solution is brought to reflux.
21.
The reaction was monitored by TLC using silica gel F
254 (200 μm thickness) glass backed plates, 1:1 EtOAc:hexanes, KMnO
4 TLC stain, (yellow spots) for the disappearance of the intermediate
N-formyl ester (R
f = 0.25), and other intermediate compounds until only the hydrochloride salt (R
f = 0.00) remains.
22.
Flash silica gel chromatography of the final product employed a column with specifications of: inner diameter: 2.5 inches; packed length: 6 inches. Fractions of ~27 mL were collected in 16 × 150 mm test tubes. Fractions containing the desired product (R
f = 0.47) were determined by TLC (90:10 CH
2Cl
2:MeOH) with KMnO
4 TLC staining. These fractions were combined and concentrated under reduced pressure (40 °C, 20-25 mm Hg).
23.
(R)-Methyl 2-allylpyrrolidine-2-carboxylate hydrochloride displays the following physical and spectral characteristics: brown oil that slowly solidifies to a brown solid (99 %
ee); optical rotation: [α]
D = -83 (
c 2, CH
2Cl
2);
1H NMR
pdf (700 MHz, CDCl
3) δ: 1.93 (bs, 1 H), 2.14 (bs, 2 H), 2.45 (bs, 1 H), 2.83-2.90 (m, 1 H), 3.03-3.10 (m, 1 H), 3.54 (bs, 1 H), 3.62 (bs, 1 H), 5.22 (d,
J = 9.8 Hz, 1 H), 5.32 (d,
J = 16.8 Hz, 1 H), 5.83-5.92 (m, 1 H), 9.55 (bs, 1 H), 10.64 (bs, 1 H);
13C NMR
pdf (176 MHz, CDCl
3) δ: 22.4, 34.5, 39.2, 45.6, 53.7, 72.5, 121.4, 130.2, 170.0; IR (thin film) 3404, 2956, 2719, 2491, 1745, 1642, 1452, 1236 cm
-1; Anal. Calcd for C
9H
16ClNO
2: C, 52.56; H, 7.84; N, 6.81; Found: C: 52.24; H: 7.69; N: 6.66.
24.
The
ee of the final product was determined via conversion of the final product to the Mosher amide using commercially available (
S)-(+)-α-methoxy-α-trifloromethylphenylacetyl chloride
(Note 25) under Schotten-Baumann conditions: In a 5-mL round-bottomed flask with a magnetic stir bar,
(R)-methyl 2-allylpyrrolidine-2-carboxylate hydrochloride (26 mg, 0.15 mmol) was partitioned between CH
2Cl
2 (0.75 mL) and water (0.75 mL). NaOH (30 mg, 0.75 mmol) was added followed by commercially available (
S)-(+)-α-methoxy-α-trifluoromethylphenylacetyl chloride (0.03 mL, 0.16 mmol). The reaction mixture was stirred open to the air for 1 h before being transferred to a 30-mL separatory funnel using CH
2Cl
2 (20 mL) and diluted with H
2O. The aqueous layer was separated and the resulting organic layer was washed with saturated NaHCO
3 (10 mL), 2 M HCl (10 mL), and
brine (10 mL). The organic phase was dried over Na
2SO
4, filtered and concentrated
in vacuo. The resulting crude product (47 mg) was analyzed by NMR spectroscopy and HPLC.
1H NMR spectroscopy of the crude material observed a single amide rotamer at room temperature. An analytical sample was obtained by purifying the crude material via flash silica gel chromatography (Inner diameter 1 cm; Packed Length 11.5 cm) eluting 2.5:97.5 to 10:90 EtOAc/hexanes to afford 25 mg of the Mosher amide.
1H NMR
pdf (700 MHz, CDCl
3) δ: 1.59-1.71 (m, 2 H), 1.88 (ddd,
J = 13.3, 7.0, 4.9 Hz, 1 H), 2.03 (ddd,
J = 13.3, 9.8, 7.0 Hz, 1 H), 2.83 (dd,
J = 14.0, 7.0 Hz, 1 H), 3.07 (ddd,
J = 11.2, 7.0, 4.2 Hz, 1 H), 3.18 (dd,
J = 14.0, 7.7 Hz, 1 H), 3.32 (ddd,
J = 11.2, 8.4, 6.3 Hz, 1 H), 3.71 (s, 3 H), 3.79 (s, 3 H), 5.15 (dd,
J = 9.8, 0.7 Hz, 1 H), 5.20 (d,
J = 16.8 Hz, 1 H), 5.81 (ddt,
J 17.5, 9.8, 7.0 Hz, 1 H), 7.40-7.41 (m, 3 H), 7.58-7.60 (m, 2 H);
13C NMR
pdf (176 MHz, CDCl
3) δ: 24.0, 34.1, 37.8, 48.7, 52.2, 55.4, 69.7, 84.5 (q,
J = 25.0 Hz), 119.7, 123.5 (q,
J = 290.4 Hz), 127.3, 127.9, 129.3, 132.8, 132.9, 164.4, 173.5;
19F NMR
pdf (376 MHz, CDCl
3) δ: -69.8; HPLC purity of the amide was determined by dissolving a sample in CH
3CN and passing it through a Phenomenex Luna 3 micron particle size C18 column (Length 100 mm; Diameter 4.6 mm) using a 60:40 solution of 0.1 % TFA in H
2O and 0.01% TFA in CH
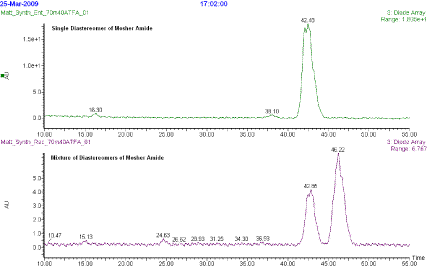
3CN at 1 mL/min over 70 min. The desired product was observed as a single peak at 42.48 min (>99% pure) that was compared to a mixture of both Mosher amide diastereomers
(Note 26). See attached chromatograph below.
25.
(
S)-(+)-α-Methoxy-α-trifloromethylphenylacetyl chloride (>99.5%
ee) was purchased from Aldrich and used without further purification.
26.
The amine L-proline methyl ester hydrochloride was protected as the
tert-butyloxycarbamate using di-
tert-butyl dicarbonate and triethylamine. Subsequent alkylation and epimerization was accomplished by deprotonating with lithium diisopropylamide followed by addition of allyl bromide. The amine was liberated by treatment with trifluoroacetic acid. The amine was converted to the Mosher amide under Schotten-Baumann conditions.
The procedures in this article are intended for use only by persons with prior training in experimental organic chemistry. All hazardous materials should be handled using the standard procedures for work with chemicals described in references such as "Prudent Practices in the Laboratory" (The National Academies Press, Washington, D.C., 2011 www.nap.edu). All chemical waste should be disposed of in accordance with local regulations. For general guidelines for the management of chemical waste, see Chapter 8 of Prudent Practices.
These procedures must be conducted at one's own risk. Organic Syntheses, Inc., its Editors, and its Board of Directors do not warrant or guarantee the safety of individuals using these procedures and hereby disclaim any liability for any injuries or damages claimed to have resulted from or related in any way to the procedures herein.
3. Discussion
As such, we have found that 2,2,2-trichloro-1-ethoxyethanol, which is commercially available, can be used as a chloral synthon resulting in a scalable procedure for the synthesis of the oxazolinone 6. In addition, we have discovered that the initial opening of the lactone to the N-formyl methyl ester intermediate is slow when using refluxing HCl in methanol. By employing the one-pot procedure described, exposure of the alkylated product (cf. 7) to sodium methoxide results in rapid conversion to the N-formyl methyl ester at room temperature. This compound is much more amenable to cleavage of the N-formyl group under refluxing HCl in methanol to reproducibly afford the desired R-allyl prolinate hydrochloride salt on a multigram scale.
Copyright © 1921-, Organic Syntheses, Inc. All Rights Reserved