Org. Synth. 1983, 61, 134
DOI: 10.15227/orgsyn.061.0134
PREPARATION OF THIOL ESTERS: S-tert-BUTYL CYCLOHEXANECARBOTHIOATE AND S-tert-BUTYL 3α,7α,12α-TRIHYDROXY-5β-CHOLANE-24-THIOATE
[Cyclohexanecarbothioic acid, S-(1,1-dimethylethyl)ester and cholane-24-thioic acid, 3,7,12-trihydroxy-S-(1,1-dimethylethyl)ester, (3α,5β,7α,12α)]
Submitted by Gary O. Spessard
1, Wan Kit Chan
2, and S. Masamune
2.
Checked by Trina Kittredge and Robert V. Stevens.
1. Procedure
Caution! Thallium compounds are very toxic. However, they may be safely handled if prudent laboratory practices are followed. Rubber gloves and laboratory coats should be worn, and reactions should be carried out in an efficient hood. Thallium wastes should be collected and disposed of separately.3
A. Thallium(I) 2-methylpropane-2-thiolate. A 500-mL, round-bottomed flask equipped with a magnetic stirring bar and a pressure-equalizing dropping funnel to which a nitrogen inlet adapter is attached is charged with 47.2 g (0.189 mol) of thallium(I) ethoxide (Note 1) and 200 mL of anhydrous benzene (Note 2). Over a period of 15 min 19.2 g (24 mL, 0.213 mol) of 2-methylpropane-2-thiol (Note 1) is added. The reaction mixture is stirred under a nitrogen atmosphere for 1 hr and the resulting precipitate is collected by filtration. After washing with three 100-mL portions of anhydrous pentane (Note 3), 48.5–51.2 g (90–95%) of the product is obtained as bright yellow crystals, mp 165–170°C dec (Note 4). This material is sufficiently pure for use in the following steps.
B. S-tert-Butyl cyclohexanecarbothioate. A solution of 4.38 g (0.030 mol) of cyclohexanecarboxylic acid chloride (Note 5) in 150 mL of ether (Note 6) is placed in a dry, 500-mL, round-bottomed flask equipped with a magnetic stirring bar and a gas inlet. The system is flushed with nitrogen and the solution is cooled in an ice bath. Stirring is initiated and 8.82 g (0.031 mol) of the thallium(I) 2-methylpropane-2-thiolate prepared in Step A is added. After the resulting milky suspension is stirred for 2 hr at room temperature, the fine precipitate is removed by filtration through Celite (Note 7) and washed thoroughly with four 50-mL portions of ether. The combined filtrate and washings are concentrated on a rotary evaporator to give a pale-yellow oil, which is distilled under reduced pressure through a 5-cm Vigreux column. After separation of a forerun, 5.36–5.44 g (90–91%) of the colorless thiol ester is collected, bp 100°C (7 mm) (Note 8).
C.
S-tert-Butyl ester from cholic acid. A
dry, 250-mL, one-necked, round-bottomed flask is equipped with a magnetic stirring bar and a nitrogen inlet adapter; the system is purged with, and maintained under, dry
nitrogen. After
4.90 g (0.0120 mol) of cholic acid (Note 9),
1.33 g (0.0131 mol) of triethylamine (Note 10), and
60 mL of dry tetrahydrofuran (THF,
(Note 11)) are placed in the flask, a stoppered, pressure-equalizing dropping funnel charged with a solution of
2.18 g (0.0127 mol) of diethyl phosphorochloridate (Note 9) in
30 mL of dry THF is attached to the top of the nitrogen inlet adapter (see
Figure 1). The solution is added to the stirred reaction mixture over a period of 5 min and stirring is continued for 3.5 hr at room temperature. The dropping funnel is removed, and the reaction mixture is taken up into a
dry, 100-mL syringe and transferred to a dry filtering apparatus. This apparatus is shown in
Figure 2. The
glass-fritted filter funnel of medium porosity with a built-in vacuum adapter is connected to the middle neck of a
500-mL, three-necked, round-bottomed flask. A
calcium chloride drying tube is connected to the
vacuum adapter and a nitrogen inlet adapter is attached to the top of the filter funnel. The precipitated
triethylamine hydrochloride is now removed from the reaction mixture by stoppering the nitrogen inlet adapter and using the positive
nitrogen pressure to force the solution through the glass frit. Dry
tetrahydrofuran, 40 mL, is used to rinse the original reaction flask. The stopper of the nitrogen inlet adapter
(Figure 2) is removed, and this washing is transferred via the same syringe to the filtering apparatus and forced through the filter in the same manner described above. One of the stoppers of the three-necked flask is replaced by a nitrogen inlet adapter and the filter funnel is replaced by a
mechanical stirrer. As the filtrate is stirred at room temperature, the remaining stopper is removed and
3.90 g (0.0133 mol) of thallium(I) 2-methylpropane-2-thiolate is added. After the addition is complete, the neck is restoppered, and the resulting mixture is vigorously stirred under
nitrogen at room temperature overnight. The precipitate is removed by suction filtration through Celite filter aid
(Note 7) and washed with three
30-mL portions of THF. The filtrate and washings are combined and concentrated under reduced pressure, and the resulting residue is dissolved in
160 mL of ethyl acetate. This solution is washed with two
100-mL portions of aqueous 5% NaHCO3, then with
50 mL of aqueous saturated NaCl, and finally is dried over anhydrous
Na2SO4. The solvent is removed by rotary evaporator to afford a white, gummy paste which crystallizes upon trituration with
20 mL of acetonitrile. The crystals are collected by suction filtration to afford 4.2 g of crude product. Recrystallization from
90 mL of hot acetonitrile provides 3.5 g of the thiol ester as small white needles, mp
166–167°C (Note 12). A second crop of 0.5 g, mp
165–166°C, can be obtained upon concentration of the mother liquor to approximately 30 mL, for a combined yield of 70%.
Figure 1
Figure 2
2. Notes
1.
Thallium(I) ethoxide and 2-methylpropane-2-thiol were purchased from Aldrich Chemical Company, Inc.2.
Benzene, reagent grade, was purified and dried by first removing the benzene–water azeotrope by simple distillation and then collecting the remaining liquid under an atmosphere of
nitrogen.
3.
Dry
pentane was obtained by allowing
practical grade pentane to be shaken with and then distilled from concentrated
sulfuric acid.
4.
The product should be stored in a
dark bottle under an atmosphere of
argon to prevent discoloration and possible decomposition.
5.
Cyclohexanecarboxylic acid chloride may be prepared in the following way: a pressure-equalizing addition funnel fitted with a
nitrogen inlet tube is attached to a 500-mL, round-bottomed flask equipped with a magnetic stirring bar and also charged with
12.8 g (0.100 mol) of cyclohexanecarboxylic acid (purchased from Aldrich Chemical Company, Inc.) and
250 mL of anhydrous ether. (Anhydrous
benzene may also be used.) The ethereal solution is cooled to
ice-bath temperature and
25.4 g (0.200 mol) of oxalyl chloride (purchased from Aldrich Chemical Company, Inc.) is added over a period of 20 min. Under
nitrogen, the resulting solution is stirred for 26 hr before it is concentrated on a rotary evaporator to afford a pale-yellow oil. Distillation of the oil yields
13.5 g (
92%) of
cyclohexanecarboxylic acid chloride as a clear, colorless liquid, bp
75°C (30 mm); IR (liquid film) cm
−1: 1800 (strong).
6.
Anhydrous ether was obtained from Mallinckrodt Inc. and used without further purification.
7.
Celite (C-211), purchased from Fisher Scientific Company, was washed thoroughly with
ether.
8.
The spectral characteristies of the product are as follows: IR (liquid film) cm
−1: 1675 (strong);
1H NMR (neat) δ: 1.42 [s, 9 H, C(C
H3)
3], 1.0–2.0 (m, 10 H, all C
H2 in cyclohexane portion), 2.3 (m, 1 H, C
H).
9.
Cholic acid and diethyl phosphorochloridate were obtained from Aldrich Chemical Company, Inc.10.
Triethylamine was purchased from Eastman Organic Chemicals.
11.
Tetrahydrofuran, reagent grade, was refluxed over and distilled from
lithium aluminum hydride immediately prior to use (see
Org. Synth., Coll. Vol. V 1973, 976 for warning).
12.
The spectral properties of the product are as follows: IR (CHCl
3) cm
−1: 3600 (sharp, weak), 3430 (broad, medium), 1675 (strong), no absorption at 1700.
3. Discussion
Methods available before 1971 for the preparation of thiol esters are briefly summarized in a review article.
4 Since then, several newer techniques have been developed to meet a certain set of criteria required for recent synthetic operations. This development may be summarized as follows. Whenever an acid chloride is available, the reaction of the Tl(I) salt of a thiolate of virtually any kind, including alkane-, benzene-, 2-benzothiazoline-, and 2-pyridinethiol, proceeds efficiently and near-quantitatively. However, if selective thiol ester formation in the presence of hydroxy or other functional groups in the same molecule is required, three main procedures are available. First, reaction of an acid (
1), with a dialkyl or diphenyl phosphorochloridate affords the anhydride (
2) (with the hydroxy groups intact) which is subsequently converted to the thiol ester.
5 This method can be applied to any type of thiol and a variety of hydroxy acids (except for β-hydroxy acids
6 7). A mixed anhydride method using
ethyl chloroformate and
pyridine also effects selective thiol ester formation in many cases.
8 Second, the imidazolide of an acid that is prepared from
1 and
N,N-carbonyldiimidazole reacts efficiently with relatively acidic thiols such as
benzenethiol to yield the thiol ester.
6,7,11 Third, use of a disulfide and
triphenylphosphine effects the selective formation of thiol esters, but this technique is applicable only to relatively reactive disulfides such as those derived from
2-benzothiazole-,
2-pyridinethiol,
9,10,12 and
4-tert-butyl-N-isopropylimidazole-2-thiol.
13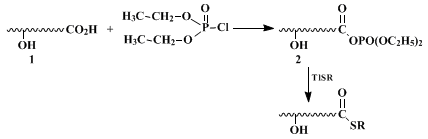
Other methods that can be used to prepare thiol esters from carboxylic acids include the use of aryl thiocyanates,
14 thiopyridyl chloroformate,
15 2-fluoro-N-methylpyridinium tosylate,
16 1-hydroxybenzotriazole,
17 and
boron thiolate.
18 Direct conversion of
O-esters to
S-esters can also be effected via
aluminum and
boron reagents.
19 20 21 However, the applicability of these
14,15,16,17,18,19,20,21 and other methods,
22 23 24 25 including the carboxyl group activation by means of
4-dimethylaminopyridine (DMAP) and
dicyclohexylcarbodiimide (DCC),
25 to the selective thiol ester formation discussed above has not been clearly defined.
Thiol esters have recently been utilized, with and without activation, for the preparation of
O-esters for lactones, in particular, in macrolide syntheses. The accompanying procedure illustrates this conversion.
26This preparation is referenced from:
Appendix
Chemical Abstracts Nomenclature (Collective Index Number);
(Registry Number)
2-benzothiazole-
boron thiolate
Celite (C-211)
sulfuric acid (7664-93-9)
Benzene (71-43-2)
ethyl acetate (141-78-6)
ether (60-29-7)
acetonitrile (75-05-8)
NaHCO3 (144-55-8)
NaCl (7647-14-5)
Na2SO4 (7757-82-6)
nitrogen (7727-37-9)
aluminum (7429-90-5)
pyridine (110-86-1)
Triethylamine hydrochloride (554-68-7)
Pentane (109-66-0)
Cyclohexanecarboxylic acid (98-89-5)
ethyl chloroformate (541-41-3)
Benzenethiol (108-98-5)
boron (7440-42-8)
Tetrahydrofuran,
THF (109-99-9)
oxalyl chloride (79-37-8)
lithium aluminum hydride (16853-85-3)
triethylamine (121-44-8)
argon (7440-37-1)
cyclohexanecarboxylic acid chloride (2719-27-9)
triphenylphosphine (603-35-0)
dicyclohexylcarbodiimide (538-75-0)
diethyl phosphorochloridate (814-49-3)
Thallium (7440-28-0)
thallium(I)
thallium(I) ethoxide (20398-06-5)
cholane-24-thioic acid, 3,7,12-trihydroxy-S-(1,1-dimethylethyl)ester, (3α,5β,7α,12α),
S-tert-BUTYL 3α,7α,12α-TRIHYDROXY-5β-CHOLANE-24-THIOATE (58587-05-6)
2-methylpropane-2-thiolate
2-methylpropane-2-thiol (75-66-1)
cholic acid
2-pyridinethiol (73018-10-7)
thiopyridyl chloroformate
1-hydroxybenzotriazole (2592-95-2)
4-dimethylaminopyridine (1122-58-3)
S-tert-BUTYL CYCLOHEXANECARBOTHIOATE,
Cyclohexanecarbothioic acid, S-(1,1-dimethylethyl)ester (54829-37-7)
N,N-carbonyldiimidazole (530-62-1)
4-tert-butyl-N-isopropylimidazole-2-thiol
2-fluoro-N-methylpyridinium tosylate (58086-67-2)
Thallium(I) 2-methylpropane-2-thiolate
Copyright © 1921-, Organic Syntheses, Inc. All Rights Reserved