Org. Synth. 1983, 61, 48
DOI: 10.15227/orgsyn.061.0048
PREPARATION OF O-ESTERS FROM THE CORRESPONDING THIOL ESTERS: tert-BUTYL CYCLOHEXANECARBOXYLATE
[Cyclohexanecarboxylic acid, 1,1-dimethylethyl ester]
Submitted by Wan Kit Chan
1, S. Masamune
1, and Gary O. Spessard
2.
Checked by Trina Kittredge and Robert V. Stevens.
1. Procedure
A 500-mL, round-bottomed flask equipped with a magnetic stirring bar is flushed with nitrogen. The flask is then charged with 5.56 g (0.028 mol) of S-tert-butyl cyclohexanecarbothioate (Note 1), 5.55 g (0.075 mol) of tert-butyl alcohol, and 250 mL of anhydrous acetonitrile (Note 2). The mixture is stirred vigorously and 23.7 g (0.056 mol) of mercury(II) trifluoroacetate (Note 3) is added in one portion. The resulting mixture is stirred vigorously for 45 min and then concentrated to approximately 50–75 mL on a rotary evaporator (Note 4). To this concentrated mixture is added 250 mL of hexane and the orange solid that forms is removed by filtration. The filter pad is then washed with 50 mL of hexane. The filtrate and washings are combined and washed with a 50 mL portion of aqueous saturated sodium chloride, dried over anhydrous sodium sulfate, and concentrated on a rotary evaporator to give a pale-yellow liquid (Note 4).
The crude product is purified by passing it through a column (4.5 cm × 30 cm) of neutral alumina (Note 5) using chloroform as eluant. The desired product moves with the solvent front, and the first 300–350 mL of eluant contains all of the product. Removal of the solvent gives 4.96 g. The product, which contains a small amount of tert-butyl alcohol, can be further purified by distillation through a short-path apparatus to give 4.6 g (90%) of pure O-ester, bp 91°C (25 mm) (Note 4) and (Note 6).
2. Notes
2.
Acetonitrile, obtained from J. T. Baker Chemical Co., was refluxed overnight with
phosphorus pentoxide and then distilled under
nitrogen onto freshly activated Linde 4A molecular sieves. The
acetonitrile was stored over the molecular sieves for 24 hr before use.
3.
Although
mercury(II) trifluoroacetate may be obtained commercially, the submitters recommend that it be freshly prepared. A mixture of
red mercury(II) oxide (108.3 g, 0.5 mol) (obtained from BDH Chemicals Ltd.) and freshly distilled
trifluoroacetic acid (137.0 g, 1.2 mol) (purchased from J. T. Baker Chemical Co.) was heated at 80°C for 30 min. The excess
trifluoroacetic acid and the water formed in the reaction were removed under reduced pressure. The white crystaline residue was then dried (50°C, 0.01 mm) for 48 hr to give a quantitative yield of product.
4.
The temperature of the
water bath was kept below 28°C during evaporation of the
acetonitrile.
5.
Woelm neutral
alumina, activity grade 1, (300 g) was used. The column was packed using
hexane.
6.
The spectral properties of the product are as follows: IR (neat) cm
−1: 1735 (strong);
1H NMR (CDCl
3) δ: 1.38 [singlet, 9 H, C(C
H3)
3] 1.0–2.4 (multiplet, 11 H, cyclohexane protons).
3. Discussion
In recent years much attention has been directed toward efficient ester (and lactone) formation in connection with the synthesis of naturally occurring macrolides.
3 4 5 6 7 8;
9;
10;
11 12 13;
14 15;
16 17 18 19 20 21 22 23 Four principal methods for such a reaction have emerged from these studies:
Method 1. Use of a thiophilic metal ion to activate an alkane- or arenethiol ester for nucleophilic displacement by an alcohol is applicable to both ester and lactone formation.
24Method 2. Corey's "double activation" method for lactone formation is patterned after Mukaiyama's procedure for peptide formation and involves refluxing a solution of the 2-pyridinethiol ester of a hydroxy acid in a high-boiling solvent for a prolonged period of time.
25Method 3. Gerlach's modification of Method 2 uses
AgClO4 or AgBF4 to catalyze the cyclization.
26Method 4. Mitsunobu's method uses a combination of
diethyl azodicarboxylate and
triphenylphosphine as a condensing agent.
27Method 1 offers some distinct advantages. First, an ester such as the 1,1-dimethyl-ethylthiol ester serves as an excellent protective group, surviving both relatively mild alkaline and acid conditions, and has been used successfully in the synthesis of many macrolide natural products.
7,12,15,19,20,22 Second, reaction of a metal ion such as Hg(II) with the thiol ester formally creates a highly reactive trivalent sulfur species, and thus ester (and lactone) formation proceeds very rapidly at room temperature or below. More importantly, bulky substituents or double bonds located near the reaction centers (i.e., near the hydroxy and acyl groups) do not impede the reaction (see Table I).
15 Thus
tert-
TABLE I
|
|
|
|
R1
|
Reagent
|
Buffer
|
Yield (%)a
|
|
|
c-C6H11-
|
1 or 2
|
Na2HPO4 (or none)
|
100
|
|
(CH3)3C-
|
1
|
|
90
|
|
(E)-CH3CH=CH-
|
1 or 2
|
|
85
|
|
(Z)-CH3CO2C(CH3)=CH-
|
1
|
Na2HPO4
|
90
|
|
aThe yields are estimated by GC analysis.
|
butyl pivalate and
tert-butyl crotonate are prepared in excellent yields. In the absence of alcohols,
tert-butyl cyclohexanecarbothioate reacts with
Hg(CF3CO2)2 to form
cyclohexanecarboxylic trifluoroacetic anhydride. Reaction of this anhydride with
tert-butyl alcohol to give the ester, however, proceeds ca. 10 times more slowly than the Hg(II)-catalyzed ester formation described above.
28 The intermediacy of the corresponding ketene has been eliminated by use of an appropriately deuterated compound
28 and
pivalic acid. Thus the metal-catalyzed ester formation appears to proceed for the most part through coordination of the alcohol to the metal, as shown in a possible intermediate (
1), followed by collapse into the ester and mercuric salts with retention of stereochemistry at the
carbon atom alpha to the carboxy group.
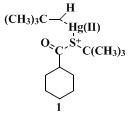
This method is not free from disadvantages: the electrophilicity of Hg(II) toward reactive alkenes may sometimes be a problem. However, in most cases the reactivity of Hg(II) with
sulfur significantly exceeds that with ordinary or electron-deficient (C=C-C=O) double bonds, and other combinations of thiol esters and thiophilic metals may be used to overcome this problem. The more acidic the reacting thiol, the less thiophilic is the metal needed to effect the reaction, and in some cases Cu(I), Cu(II), and Ag(I) are superior to Hg(II). For example, the combination of Ag(I)CF
3CO
2 [but not Ag(I)ClO
4 or Ag(I)BF
4] and a benzenethiol ester is very efficient for ester formation. The presence of electron-withrawing groups such as the C=C bond and protected hydroxy groups somewhat retards the ester formation. A few examples are shown in Table II.
15 All these observations appear to conform with the hard and soft acid and base principle of Pearson. Further, it is clear that Gerlach's report of the use of Ag(I) to activate 2-pyridinethiol esters (Method 3) is fully in accord with this trend.
Corey's "double activation" procedure (Method 2) does not use an external reagent to activate the functional group, but effects cyclization by heating a solution of the 2-pyridinethiol ester of a hydroxy acid for a prolonged period. Several pieces of evidence point to the intermediacy of
2 in this lactonization.
29 If one accepts this intermediate, it follows that a hydroxy(2-pyridinethiol)ester, heavily substituted near the reaction centers (i.e., near the hydroxyl and acyl groups), would encounter a high energy barrier in the process leading to
2. This inference has been confirmed by measuring the approximate rates of reaction of 2-pyridine- and 2-benzothiazolethiol esters of
cyclohexanecarboxylic acid with primary, secondary, and tertiary alcohols.
3,28 This steric retardation of the reaction may constitute a major drawback to Method 2. A marked improvement has been made, however, and the latest version of this method
30 involves use of the 1-methyl- or 1-isopropyl-4-
tert-butylimidazole-2-thiol ester of the hydroxy acid which undergoes cyclization ~100 times faster than the corresponding 2-pyridinethiol ester. This improved method has been used in syntheses of erythronolide A
18 and B.
16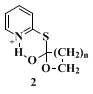
Lactonization of aliphatic hydroxy acids proceeds with the aid of two reagents,
diethyl azodicarboxylate and
triphenylphosphine. This fourth procedure has been selected as a method of choice for the final cyclization to yield
vermiculine14 and
pyrenophorin.
13Several other methods to effect ester and lactone formation are now available. Mukaiyama uses
2-chloro-N-methylpyridinium iodide and its derivatives as a condensing agent.
31;
32;
33;
34;
35 Staab's imidazole method,
36 successfully utilized in a synthesis of
pyrenophorin8 and a model study for
erythronolide B,
37 requires a catalytic amount of strong base, and thus is applicable only to compounds stable under such conditions. The mixed anhydride of a
hydroxycarboxylic acid and
2,4,6-trichlorobenzoic acid is efficiently cyclized to provide the corresponding lactone.
37,38;
39 Similarly, the use of a reactive phosphoric acid anhydride intermediate is equally effective.
40 Some other methods for carboxyl activation, using
dibutyltin oxide,
41;
42 distannoxane,
43 N,N,N',N'-tetramethychloroformamidinium chloride,
44 and the combination of
dicyclohexylcarbodiimide (DCC),
4-dimethylaminopyridine (DMAP), and
DMAP hydrochloride,
45 have also appeared.
TABLE II
|
|
|
|
R1
|
R2
|
MX
|
Solvent
|
Time
|
Yield (%)
|
|
|
c-C6H11-
|
-C6H5
|
Cu(CF3SO3)
|
C6H6/THF
|
10 min
|
95
|
|
c-C6H11-
|
|
Ag(CF3CO2)
|
C6H6
|
10 min
|
100
|
|
(E)-CH3CH=CH-
|
-C6H5
|
Cu(CF3SO3)
|
C6H6/THF
|
5 hr
|
80
|
|
(E)-C6H5=CH-
|
-C6H5
|
Cu(CF3SO3)2
|
CH3CN
|
1.5 hr
|
24
|
|
Ag(CF3CO2)
|
C6H6(Δ)
|
1.5 hr
|
100
|
|
AgBF4
|
C6H6(Δ)
|
1 hr
|
<5
|
|
C6H5-
|
|
Ag(CF3CO2)
|
C6H6(Δ)
|
1.5 hr
|
100
|
|
Cu(CF3SO3)
|
C6H6/THF
|
5 hr
|
90
|
|
Cu(CF3SO3)2
|
CH3CN
|
30 min
|
100
|
|
The preceding discussion is a summary of the lactonization methods known at present; newer methods continue to be explored. The selection of a method for an individual case depends to a large extent on the structure and functionalities of the substrate.
This preparation is referenced from:
Appendix
Chemical Abstracts Nomenclature (Collective Index Number);
(Registry Number)
alumina
red mercury(II) oxide
AgClO4
AgBF4
Na2HPO4
Hg(CF3CO2)2
cyclohexanecarboxylic trifluoroacetic anhydride
vermiculine
pyrenophorin
erythronolide B
N,N,N',N'-tetramethychloroformamidinium chloride
DMAP hydrochloride
acetonitrile (75-05-8)
chloroform (67-66-3)
sodium chloride (7647-14-5)
sodium sulfate (7757-82-6)
nitrogen (7727-37-9)
sulfur (7704-34-9)
carbon (7782-42-5)
Cyclohexanecarboxylic acid (98-89-5)
Pivalic acid (75-98-9)
diethyl azodicarboxylate (1972-28-7)
hexane (110-54-3)
hydroxycarboxylic acid (463-79-6)
tert-butyl alcohol (75-65-0)
trifluoroacetic acid (76-05-1)
triphenylphosphine (603-35-0)
dicyclohexylcarbodiimide (538-75-0)
mercury(II) trifluoroacetate (13257-51-7)
4-dimethylaminopyridine (1122-58-3)
Cyclohexanecarboxylic acid, 1,1-dimethylethyl ester,
tert-Butyl cyclohexanecarboxylate (16537-05-6)
butyl pivalate
2,4,6-trichlorobenzoic acid (50-43-1)
dibutyltin oxide (818-08-6)
distannoxane
phosphorus pentoxide (1314-56-3)
S-tert-BUTYL CYCLOHEXANECARBOTHIOATE (54829-37-7)
tert-butyl crotonate (79218-15-8)
tert-butyl cyclohexanecarbothioate
2-chloro-N-methylpyridinium iodide (14338-32-0)
Copyright © 1921-, Organic Syntheses, Inc. All Rights Reserved