Org. Synth. 2023, 100, 159-185
DOI: 10.15227/orgsyn.100.0159
Synthesis of a 2,5-Bis(tert-butyldimethylsilyloxy)furan and its Reaction with Benzyne
Submitted by Jessica E. Budwitz and Christopher G. Newton*
1Checked by Dave J. Charboneau, Jordan K. Thompson, and Sarah E. Reisman
1. Procedure (Note 1)
A. ((3-Methylfuran-2,5-diyl)bis(oxy))bis(tert-butyldimethylsilane) (1). A 1000 mL, three-necked (14/20, 29/42, 14/20), round-bottomed flask is equipped with a 16 × 38 mm, egg-shaped, Teflon-coated magnetic stir bar, two 14/20 glass stoppers, and a 29/42 glass gas adapter with a gas inlet (Figure 1A). The apparatus is flame-dried under vacuum and purged with inert gas in triplicate (Note 2). The setup is maintained under a slight positive pressure of inert gas and allowed to cool to ambient temperature (22 °C). The flask is charged with methylsuccinic anhydride (10.0 g, 87.6 mmol, 1 equiv) (Note 3), and a glass stopper is replaced with a 14/20 rubber septum. Diethyl ether (350 mL, 0.25 M) (Note 4) is transferred into the flask via cannula (18 gauge), and vigorous stirring (700 rpm) is initiated. Once methylsuccinic anhydride is fully dissolved (ca. 5 min, colorless solution), the remaining glass stopper is replaced with a 14/20 Teflon thermometer adapter equipped with a glass thermometer (range of -20 °C to 250 °C) (Figure 1B). The reaction flask is submerged in an ice/water bath and cooled for 10 min to an internal temperature of 0 °C. Triethylamine (42.8 mL, 306.7 mmol, 3.5 equiv) (Note 5) is added to the flask in one portion using a 60 mL syringe. tert-Butyldimethylsilyl trifluoromethanesulfonate (50.3 mL, 219.1 mmol, 2.5 equiv) (Note 6) is added dropwise over 10 min using a 60 mL syringe, ensuring an internal temperature of 5 °C is not exceeded. Formation of a white cloudy precipitate and mild fuming is observed (Figure 1C). The precipitate dissolves during the course of the addition, and fumes dissipate shortly after addition is complete, resulting in a cloudy, colorless, biphasic mixture. The ice/water bath is removed, the reaction is allowed to warm to ambient temperature, and both the glass thermometer and septum are replaced with 14/20 glass stoppers (Figure 1D).

Figure 1. (A) Setup for procedure A; (B) Appearance of reaction after dissolution of methylsuccinic anhydride; (C) Appearance of reaction during tert-butyldimethylsilyl trifluoromethanesulfonate addition; (D) Appearance of reaction 20 minutes after tert-butyldimethylsilyl trifluoromethanesulfonate addition (photos provided by submitters)
Completion of the reaction is confirmed by 1H NMR analysis (Notes 7 and 8), by which time the reaction is a bright orange/yellow color (Figures 2A and 2B). The crude mixture is filtered through a 600 mL, 10.5 cm diameter sintered funnel (medium grit) packed with 150 g of phosphate buffered silica (pH = 7, topped with filter paper) (Notes 9, 10, 11, and 12) eluting with diethyl ether (Note 13) until all product has eluted (determined by TLC) into a 1000 mL, single-necked (24/40), round-bottomed flask to remove the insoluble orange triflate salts (Figure 2C). The round-bottomed flask is placed on a rotary evaporator, and the solvent and triethylamine are removed (300-22 mmHg at 35 °C), providing the crude product as a yellow oil. The crude product is transferred to a 100 mL, single-necked (14/20), round-bottomed flask (Figure 2D) (Notes 14 and 15).

Figure 2. (A) Appearance of reaction after reaching completion (with stirring); (B) Appearance of reaction after reaching completion (without stirring); (C) Filtration over phosphate buffered silica; (D) Crude ((3-methylfuran-2,5-diyl)bis(oxy))bis(tert-butyldimethylsilane) (1) (photos provided by submitters)
The round-bottomed flask containing the crude product is equipped with a 4.7 × 9.5 mm, egg-shaped, Teflon-coated magnetic stir bar and an oven-dried 14/20 one-piece short path vacuum distillation apparatus equipped with a 10/14 glass stopper and a 10 mL, single-necked (14/20), round-bottomed flask (Figure 3A). Chilled water circulation and stirring (500 rpm) are initiated, the setup is placed under vacuum (ca. 2.50 mmHg), and the distillation apparatus is insulated with glass wool and aluminum foil (Figure 3B). The crude mixture is warmed on a heating mantle at 80 °C to remove the majority of volatile impurities (Note 16). This process takes approximately 1 h, and several mL of distillate are collected. Once distillate collection in the receiving flask has ceased, the round-bottomed flask is cooled to ambient temperature and placed under a slight positive pressure of inert gas (Note 2). The distillation apparatus is replaced with a 14/20 glass gas adapter and the setup is re-exposed to vacuum. The round-bottomed flask is submerged in a water bath at 80 °C (Figure 3C). The flask is heated for several hours to remove the remaining traces of volatile impurities (Note 17), providing the crude product as a yellow oil. The crude residue is subjected to flash column chromatography with phosphate buffered silica (pH = 7) (Notes 18 and 19) to give ((3-methylfuran-2,5-diyl)bis(oxy))bis(tert-butyldimethylsilane) (1) (22.2 g, 74% yield, 97% purity) as a clear, colorless oil (Notes 20, 21, and 22) (Figure 3D).

Figure 3. (A) Setup for distillation. (B) Removal of volatile impurities by distillation. (C) Appearance of crude mixture following distillation. (D) Pure ((3-methylfuran-2,5-diyl)bis(oxy))bis(tert-butyldimethylsilane) (1) (photos provided by submitters)
B. 2-Iodophenyl trifluoromethanesulfonate (2). A 500 mL, three-necked (24/40, 24/40, 24/40), round-bottomed flask is equipped with a 9.5 ´ 19 mm, egg-shaped, Teflon-coated magnetic stir bar, two 24/40 glass stoppers, and a 24/40 glass gas adapter with a gas inlet (Figure 4A). The apparatus is flame-dried under vacuum and purged with inert gas in triplicate (Note 2). The setup is maintained under a slight positive inert gas pressure and allowed to cool to ambient temperature (22 °C). The flask is charged with 2-iodophenol (15.0 g, 68.2 mmol, 1 equiv) (Note 23), and a glass stopper is replaced with a 24/40 rubber septum. Dichloromethane (134 mL, 0.5 M) (Note 24) is transferred into the flask via cannula (18 gauge), then stirring (500 rpm) is initiated, affording a transparent brown solution. The remaining glass stopper is replaced with a 24/40 Teflon thermometer adapter equipped with a glass thermometer (range of -20 °C to 250 °C) (Figure 4B). The reaction flask is submerged in an ice/water bath and cooled to 0 °C (internal reaction temperature). This process takes approximately 10 min. Pyridine (11.0 mL, 136.4 mmol, 2 equiv) (Note 25) is added in one portion using a 20 mL syringe, resulting in formation of a bright orange solution and an increase in internal reaction temperature to 6 °C) (Figure 4C). The reaction is allowed to cool to 0 °C, at which time trifluoromethanesulfonic anhydride (14.4 mL, 85.2 mmol, 1.25 equiv) (Note 26) is added dropwise using a 20 mL syringe at a rate that ensures an internal reaction temperature of 10.0 °C is not exceeded. The addition process takes approximately 20 minutes, and formation of a fine, white precipitate accompanied by dense, white, cloudy fumes are observed. The ice/water bath is removed, and the reaction is allowed to warm to ambient temperature, during which time white fumes dissipate (Figure 4D).

Figure 4. (A) Setup for procedure B; (B) Appearance of reaction after dichloromethane addition; (C) Appearance of reaction after pyridine addition; (D) Appearance of reaction 20 minutes after removal of ice/water bath (photos provided by submitters)
The reaction is complete by the time the solution has warmed to ambient temperature (ca. 20 minutes after the removal of the ice/water bath). Completion of the reaction is confirmed by TLC (Note 27). The reaction mixture is diluted with deionized water (300 mL) and transferred to a 1000 mL separatory funnel, rinsing the reaction flask with dichloromethane (Note 28) and deionized water. The organic layer is removed, and the aqueous layer is extracted with dichloromethane (2 × 100 mL). The organic layers are combined and washed with 2 M HCl (2 × 300 mL), saturated aqueous sodium bicarbonate (1 × 300 mL), deionized water (1 × 300 mL), and brine (1 × 300 mL). The organic layer changes color from bright orange to orange-brown during the HCl wash, then returns to a bright orange color when washed with saturated aqueous sodium bicarbonate. The organic layer is dried over MgSO4 (13.0 g) and filtered through a 150 mL, 7.5 cm diameter sintered funnel (medium grit) into a 1000 mL, single-necked (24/40), round-bottomed flask. The flask is placed on a rotary evaporator, and the solvent is removed (180-22 mmHg at 25 °C), providing the crude product as a bright orange oil (Figure 5A). The crude residue is subjected to flash column chromatography on silica gel (Note 29) using hexanes and ethyl acetate (Notes 19 and 30) to give 2-iodophenyl trifluoromethanesulfonate (2) (23.10 g, 96% yield, 99% purity) as a colorless oil (Notes 31 and 32) (Figure 5B).

Figure 5. (A) Crude 2-iodophenyl trifluoromethanesulfonate (2); (B) Pure 2-iodophenyl trifluoromethanesulfonate (2) (photos provided by submitters)
C. 2-Methylnaphthalene-1,4-dione (3). A 500 mL, three-necked (24/40, 24/40, 24/40), round-bottomed flask is equipped with a 9.5 × 19 mm, egg-shaped, Teflon-coated magnetic stir bar, two 24/40 glass stoppers, and a 24/40 glass gas adapter with a gas inlet (Figure 6A). The apparatus is flame-dried under vacuum and purged with inert gas, a process that is performed in triplicate (Note 2). The setup is maintained under a slight positive inert gas pressure and allowed to cool to ambient temperature (22°C). A glass stopper is replaced with a 24/40 rubber septum, and the flask is charged with ((3-methylfuran-2,5-diyl)bis(oxy))bis(tert-butyldimethylsilane) via syringe transfer (1) (18.3 g, 19.6 mL, 53.3 mmol, 1.25 equiv) and 2-iodophenyl trifluoromethanesulfonate (2) (15.0 g, 7.74 mL, 42.6 mmol, 1 equiv) via syringe transfer. Both 1 and 2 are degassed prior to addition through exposure to vacuum at room temperature for 5 min before transferring to a syringe under a positive pressure of inert gas. A positive pressure of inert gas is maintained throughout the syringe transfer. Tetrahydrofuran (171 mL, 0.25 M) (Note 33) is transferred into the flask via cannula (18 gauge), and the remaining glass stopper is exchanged for a 24/40 Teflon thermometer adapter equipped with an ultra-low temperature glass thermometer (range of -100 to 50 °C) (Figure 6B). Stirring is initiated (650 rpm), and the reaction flask is submerged in an acetone/dry ice bath and cooled to -70 °C (internal reaction temperature), a process requiring approximately 30 minutes. n-Butyllithium (1.23 M solution in hexanes, 43.3 mL, 53.3 mmol, 1.25 equiv) (Note 34) is added via syringe pump with a 60 mL syringe over 20 min (initial increase in internal reaction temperature to -58 °C). At the midpoint of the addition the reaction is a light pink color (Figure 6C). Following completion of n-butyllithium addition (internal reaction temperature of -64 °C), the reaction is maroon in color (Figure 6D).
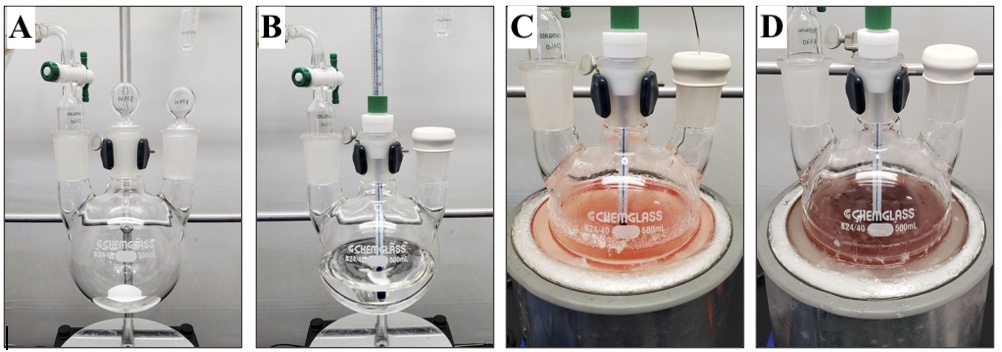
Figure 6. (A) Setup for procedure A; (B) Appearance of reaction after tetrahydrofuran addition; (C) Appearance of reaction midway through n-butyllithium addition; (D) Appearance of reaction immediately after n-butyllithium addition (photos provided by submitters)
The reaction is stirred for a further 10 min then quenched by dropwise addition (ca. 1 min) of saturated aqueous ammonium chloride solution (10 mL) via syringe. A small exotherm (-54 °C) and formation of a white precipitate are observed (Figure 7A). The reaction is allowed to warm to ambient temperature (ca. 1 h), at which point the reaction mixture is bright yellow with precipitate still present (Figure 7B). The reaction mixture is transferred to a 2000 mL separatory funnel, with subsequent rinsing of the reaction flask with ethyl acetate (100 mL) (Note 30) and deionized water (100 mL). The mixture is further diluted with 450 mL of deionized water to dissolve the precipitate, and the mixture is extracted with ethyl acetate (2 × 200 mL). The combined organic layers are washed with 2 M HCl (3 × 300 mL), saturated aqueous sodium bicarbonate (1 × 300 mL), deionized water (1 × 300 mL), and brine (1 × 300 mL). The organic layer is dried over MgSO4 (24.6 g) and filtered through a 350 mL, 9.5 cm diameter sintered funnel (medium grit) into a 1000 mL, single-necked (24/40), round-bottomed flask. The flask is placed on a rotary evaporator and solvent is removed (75-22 mmHg at 25 °C) to afford a yellow solid (Figure 7C). The crude product is transferred to a 500 mL, single-necked (24/40), round-bottomed flask and residual solvent is removed under high vacuum (ca. 2.5 mmHg, ambient temperature for 3 h).

Figure 7 (A) Appearance of reaction after ammonium chloride addition; (B) Appearance of reaction once warmed to ambient temperature; (C) Crude 2-methylnaphthalene-1,4-dione (3) (photos provided by submitters)
The round-bottomed flask is equipped with a 16 × 38 mm, egg-shaped, Teflon-coated magnetic stir bar. Hot hexanes (50 mL, 60 °C) (Note 19) are transferred into the flask, and stirring (400 rpm) of the mixture is initiated. The crude mixture is warmed to reflux (Figure 8A), and heating is maintained until the crude residue completely dissolves (ca. 5 min) (Note 35). The flask is equipped with a 24/40 plastic stopper (joint wrapped with Parafilm). The flask is removed from the heating mantle and allowed to cool to ambient temperature, by which time significant crystallization has occurred (Figure 8B). The flask is transferred to a freezer (-20 °C) for 12 h, then removed and immediately filtered through a 350 mL, 9.5 cm diameter sintered funnel (medium grit), eluting with a minimum volume of cold hexanes (ca. -20 °C) to give small, bright yellow crystals. The solid is transferred to a 50 mL beaker charged with a magnetic stir bar and stirred rigorously at room temperature for 5 min in hexanes (approximately 4 mL per gram of material). The crystals are collected by filtration through a 30 mL, 3.5 cm diameter sintered funnel (medium grit), eluting with a minimum volume of hexanes (ca. 20 mL). The resulting yellow solid is dried for 3 h under high vacuum (ca 2.5 mmHg, ambient temperature) to provide 2-methylnaphthalene-1,4-dione (3) (6.18 g, 84% yield, 98% purity) (Notes 36, 37, 38, and 39) (Figure 8C).

Figure 8 (A) Appearance of crystallization midway through dissolution of crude residue; (B) Appearance of crystallization once cooled to ambient temperature; (C) Pure 2-methylnaphthalene-1,4-dione (3) (photos provided by submitters)
2. Notes
1. Prior to performing each reaction, a thorough hazard analysis and risk assessment should be carried out with regard to each chemical substance and experimental operation on the scale planned and in the context of the laboratory where the procedures will be carried out. Guidelines for carrying out risk assessments and for analyzing the hazards associated with chemicals can be found in references such as Chapter 4 of "Prudent Practices in the Laboratory" (The National Academies Press, Washington, D.C., 2011; the full text can be accessed free of charge at
https://www.nap.edu/catalog/12654/prudent-practices-in-the-laboratory-handling-and-management-of-chemical. See also "Identifying and Evaluating Hazards in Research Laboratories" (American Chemical Society, 2015) which is available via the associated website "Hazard Assessment in Research Laboratories" at
https://www.acs.org/content/acs/en/about/governance/committees/chemicalsafety/hazard-assessment.html. In the case of this procedure, the risk assessment should include (but not necessarily be limited to) an evaluation of the potential hazards associated with
methylsuccinic anhydride,
diethyl ether,
triethylamine,
tert-butyldimethylsilyl trifluoromethanesulfonate,
deuterated chloroform,
potassium carbonate, deionized water,
sodium phosphate dibasic, silica gel,
phosphoric acid,
hexanes,
2-iodophenol,
dichloromethane,
pyridine,
trifluoromethanesulfonic anhydride,
hydrochloric acid (2 M),
ammonium chloride (aqueous),
sodium bicarbonate (aqueous),
sodium chloride (aqueous),
magnesium sulfate,
ethyl acetate,
tetrahydrofuran,
n-butyllithium (1.6 M),
potassium permanganate, as well as the proper procedures for working under an inert atmosphere, solvent evaporation, the application of high vacuum, and working with dry ice.
2. Inert gases used include argon (submitters) or nitrogen (checkers). Argon (AR UHP300, Ultra High Purity) was purchased from Airgas and used as received. Nitrogen gas was dried via passage through a Drierite column.
3.
Methylsuccinic anhydride (98%) was purchased from Combi-Blocks. To aid in dissolution it was ground into a powder using a mortar and pestle directly prior to use.
4.
Diethyl ether (≥99.9%, CHROMASOLV
TM Plus, inhibitor-free, for application as reaction solvent) was purchased from Honeywell and passed through an activated alumina column before use.
5.
Triethylamine (anhydrous, 99.8%, Sure/Seal
TM packaging system) was purchased from Sigma-Aldrich and used as received.
6.
tert-Butyldimethylsilyl trifluoromethanesulfonate (98%) was purchased from Oakwood Chemical and used as received.
7. Stirring of the reaction mixture is halted, and a 0.1 mL aliquot of the top layer is withdrawn via syringe. The aliquot is transferred to a 10 mL, single-necked (14/20), round-bottomed flask, and the majority of
diethyl ether is removed by rotary evaporation (225 mmHg, 25 °C). The residue is dissolved in approximately 1 mL of CDCl
3 (stored over
potassium carbonate), and the solution is transferred to an NMR tube for analysis.
8. The reaction was stirred for 16 h before the first NMR aliquot was taken to confirm reaction completion. An otherwise identical reaction run on a smaller scale (2.0 g of
methylsuccinic anhydride) was observed to be complete within 6 h.
9. Preparation of phosphate buffered silica (pH = 7): A 5000 mL Erlenmeyer flask is equipped with a 9.5 × 63.5 mm, octagon, Teflon-coated magnetic stir bar and 3800 mL of deionized water is added. Stirring is initiated (600 rpm), and
sodium phosphate dibasic (113.6 g, 800 mmol) (
Note 10) is slowly added, allowing it to evenly disperse (aids dissolution). Additional deionized water is added to reach a total volume of 4000 mL (0.2 M). Once fully dissolved (ca. 10 min) (Figure 9A), 400 g of silica gel (
Note 11) is slowly added, allowing it to evenly disperse (Figure 9B). The pH of the mixture is measured (pH paper, range 1-13) to confirm that it is neutral. If basic, the mixture is neutralized by either: (i) dropwise addition of
phosphoric acid (
Note 12), or (ii) addition of further silica gel (10 g portions). If acidic, additional phosphate buffer (0.2 M, prepared as above) is added. Once neutral, the mixture is filtered through a 2000 mL, 13.5 cm diameter sintered funnel (medium grit), then air is pulled through the funnel for 30 min (Figure 9C). The phosphate buffered silica is transferred to a 2000 mL shallow glass dish (Figure 9D) and placed in an oven (105 °C) for 3 days, stirring every day to ensure even drying. The phosphate buffered silica is allowed to cool to ambient temperature (Figure 9E) and is sifted through a sieve into a container for long term storage (Figure 9F). Appearance: white, free-flowing solid that partially adheres to glass when wet with solvent. Note: when using phosphate buffered silica, the addition of acidified sand to ensure a level silica gel line should be avoided. The submitters recommend using additional phosphate buffered silica as a sand replacement.

Figure 9. (A) Appearance following complete dissolution of sodium phosphate dibasic; (B) Appearance following addition of silica gel; (C) Filtration of phosphate buffered silica; (D) Appearance of wet phosphate buffered silica; (E) Appearance of dry phosphate buffered silica; (F) Sifting of dry phosphate buffered silica (photos provided by submitters)
10.
Sodium phosphate dibasic (98%) was purchased from Oakwood Chemical and used as received.
11. Silica gel (SiliCycle
TM SiliaFlash
TM P60, 230-400 mesh, 60 Å) was purchased from Fisher Scientific and used as received.
12.
Phosphoric acid (85%) was purchased from Oakwood Chemical and used as received.
13.
Diethyl ether (≥99%, for application during reaction purification) was purchased from Avantor and used as received.
14. Compound
1 following initial filtration over buffered silica: 29.0 g, 82% yield, 85% purity. From our experiences, most
2,5-bis(tert-butyldimethylsilyloxy)furan derivatives can be employed in subsequent Diels-Alder studies without additional purification, although in these cases a larger excess of reagents is typically necessary (for representative examples, see reference
6).
15. If desired, purity can be determined by quantitative
1H NMR spectroscopy using
1,2-dichloroethane (submitters) or
1,3,5-trimethoxybenzene (checkers) as an internal standard.
16. Two volatile impurities are removed by vacuum distillation. Under the reported conditions they co-distill as a clear, colorless oil [
1H NMR (400 MHz, CDCl
3): δ 0.91, 0.86, 0.10, 0.01 ppm, Figure 10]. Traces of
1 are occasionally observed in the distillate.
Figure 10. 1H NMR analysis of volatile impurities (spectra provided by submitters)
17. The final traces of volatile impurities condense on the walls of the glass gas adapter. These can be removed by either: (i) gently warming the adaptor with a heat gun while the setup is still under vacuum. In this case it is important to ensure that any impurities that may build up in the Tygon tubing are unable to drip back into the flask by having your Tygon tubing running initially downhill from the glass inlet adaptor. (ii) Placing the system back under inert gas, removing the glass adaptor, and rinsing with
acetone. This process is repeated until the volatile impurities are no longer present in the bulk solution (determined by
1H NMR).
18. Column chromatography is performed using 130 g of phosphate buffered silica (pH = 7, see
Note 9 for preparation) in a 1000 mL, 5.1 cm diameter column. The silica is pre-wet with
hexanes, and compound
1 is loaded using a minimum volume of
hexanes. An isocratic solvent system is employed, eluting with 1000 mL of
hexanes (
Note 19). Fractions of 50 mL are collected, totaling 18 fractions. Each fraction is spotted onto a glass-backed silica gel plate and visualized with UV light to confirm the presence of
1 (the TLC plates are not developed due to the instability of
1 on silica gel plates). The desired product is obtained in fractions 6-14, and these are combined into a 1000 mL, single-necked (24/40), round-bottomed flask. The bulk of the solvent is removed by rotary evaporation (150-33 mmHg, 35 °C). The product is transferred to a 100 mL, single-necked (14/20), round-bottomed flask and residual solvent is removed under high vacuum (ca. 2.5 mmHg, ambient temperature for 3 h).
19.
Hexanes (≥98.5%, ACS grade, for application during reaction purification) was purchased from Macron Fine Chemicals and used as received.
20. The product (
1) possesses the following properties: R
f: N/A (unstable on glass-backed silica gel TLC plates); bp 124-134 °C (4.3 mmHg);
1H NMR
pdf (400 MHz, CDCl
3) δ: 4.81 (s, 1H), 1.75 (s, 3H), 0.97 (s, 9H), 0.95 (s, 9H), 0.20 (s, 6H), 0.18 (s, 6H);
13C NMR
pdf (101 MHz, CDCl
3) δ: 146.7, 143.0, 92.7, 86.7, 25.5, 25.5, 18.1, 18.0, 8.80, -4.6, -4.8; IR (NaCl, thin film): 2931, 2859, 1665, 1628, 1255, 892, 787 cm
-1; HRMS (FD
+): calcd for C1
7H3
4O
3Si
2: 342.20465. Found: 342.20481. Purity was determined to be 97% by quantitative
1H NMR
pdf using 50.0 mg of
1 and 24.5 mg of
1,3,5-trimethoxybenzene (>99%, purchased from Millipore Sigma).
21. The submitters recommend storing
1 neat under an argon atmosphere at -20 °C. Under these conditions no discernible decomposition was observed over the course of several weeks. Purity can be checked by
1H NMR.
22. A second run on identical scale yielded 21.9 g (73%, 98% purity) of product.
23.
2-Iodophenol (≥98%) was purchased from Matrix Scientific and used as received.
24.
Dichloromethane (99%, HPLC grade, for application as reaction solvent) was purchased from Fisher Scientific and passed through an activated alumina column before use.
25.
Pyridine (anhydrous, 99.8%, Sure/Seal
TM packaging system) was purchased from Sigma-Aldrich and used as received.
26.
Trifluoromethanesulfonic anhydride (99.5%) was purchased from Oakwood Chemical and used as received.
27. The progress of the reaction is evaluated using TLC analysis with glass-backed silica gel plates, developed with 10%
ethyl acetate in
hexanes and visualized with UV light. The R
f of
2-iodophenol is 0.20, and the R
f of
2-iodophenyl trifluoromethanesulfonate (
2) is 0.46.
Figure 11. TLC of reaction mixture 20 minutes after removal of ice/water bath (SM = 2-iodophenol, C = co-spot of SM/R, R = reaction mixture) (photo provided by submitters)
28.
Dichloromethane (≥99.5%, Stabilized/Certified ACS, for application during reaction purification) was purchased from Fisher Scientific and used as received.
29. Column chromatography is performed using 254 g of silica gel (
Note 11) in a 1000 mL, 5.1 cm diameter column. The silica is pre-wet using 10%
ethyl acetate (
Note 30) in
hexanes (
Note 19) and acidified sand is used to ensure a level silica gel line. Compound
2 is loaded using a minimum volume of
dichloromethane. An isocratic solvent system is employed, eluting with 1500 mL of 10%
ethyl acetate in
hexanes. Fractions of 50 mL are collected, totaling 30 fractions. The desired product is obtained in fractions 9-24, and are combined into a 1000 mL, single-necked (24/40), round-bottomed flask. The bulk of the solvent is removed by rotary evaporation (120-22 mmHg at 25 °C). The product is transferred to a 25 mL, single-necked (14/20), round-bottomed flask and residual solvent is removed under high vacuum (ca. 2.5 mmHg, ambient temperature for 3 h).
30.
Ethyl acetate (≥99.5%, Certified ACS, for application during reaction purification) was purchased from Fisher Scientific and used as received.
31. The product (
2) possesses the following properties: R
f: 0.46 (10%
EtOAc in
hexanes);
1H NMR
pdf (400 MHz, CDCl
3) δ: 7.94 (dd,
J = 7.9, 1.6 Hz, 1H), 7.46 (ddd,
J = 8.8, 7.4, 1.6 Hz, 1H), 7.36 (dd,
J = 8.3, 1.5 Hz, 1H), 7.14 (td,
J = 7.7, 1.5 Hz, 1H);
13C NMR
pdf (101 MHz, CDCl
3) δ: 150.3, 140.8, 130.1, 129.6, 122.1, 118.7 (q,
J = 320.6 Hz), 89.1; IR (thin film): 1424, 1218, 1138, 885, 739 cm
-1; HRMS (FD
+) [M]
+ calcd for C
7H
4O
3F
3SI: 351.88779. Found: 351.88806. Purity was determined to be 99% by quantitative
1H NMR
pdf using 50.0 mg of
2 and 23.9 mg of
1,3,5-trimethoxybenzene (>99%, purchased from Millipore Sigma).
32. A second run on identical scale yielded 23.4 g (97% yield, 99% purity) of product, which was pale red/orange in color.
33.
Tetrahydrofuran (≥99.9%, CHROMASOLV
TM Plus, inhibitor-free, for application as reaction solvent) was purchased from Honeywell and passed through an activated alumina column before use.
34.
n-Butyllithium (1.6 M solution in
hexanes, Sure/Seal
TM packaging system) was purchased from Sigma-Aldrich and titrated prior to use in triplicate (average of 1.23 M). Titrations were conducted according to the method of Kofron.
2 Diphenylacetic acid (98%) was purchased from Combi-Blocks and recrystallized from anhydrous
toluene prior to use. Use of a cannula to add large quantities of
n-butyllithium is generally viewed as a safer alternative.
35. It is recommended that the flask be equipped with a reflux condenser that is connected to a nitrogen line.
36. The product (
3) possesses the following properties: R
f: 0.27 (10%
EtOAc in
hexanes), visualized with
KMnO4; mp 102.7-106.2 °C;
1H NMR
pdf (400 MHz, CDCl
3) δ: 8.13 - 8.08 (m, 1H), 8.06 (ddd,
J = 5.7, 3.3, 0.6 Hz, 1H), 7.73 (dd,
J = 5.8, 3.3 Hz, 2H), 6.84 (q,
J = 1.6 Hz, 1H), 2.20 (d,
J = 1.6 Hz, 3H);
13C NMR
pdf (101 MHz, CDCl
3) δ: 185.6, 185.0, 148.2, 135.7, 133.7, 133.6, 132.3, 132.2, 126.6, 126.1, 16.5; IR (thin film): 3060, 1655, 1583, 1295, 1255, 1156, 897, 776, 657 cm
-1; HRMS (FD
+): [M]
+ calcd for C
11H
8O
8: 172.05243. Found: 172.05156. Purity was determined to be 98% by quantitative
1H NMR
pdf spectroscopy using 10.5 mg of
3 and 4.5 mg of
1,3,5-trimethoxybenzene (>99%, purchased from Sigma Aldrich).
37. A second run on half of the reported scale yielded 2.63 g (72% yield, 98% purity) of product.
38. If the yield is lower than expected, the filtrate of the recrystallization can be resubjected to the crystallization conditions by concentration of the filtrate under reduced pressure and subjection of the residue to the same crystallization procedure (3 mL of
hexanes), providing a second crop of
2-methylnaphthalene-1,4-dione (
3) (0.81 g).
39. The product (
3) was determined to be light sensitive. A 10 mg sample of 3 (purchased from Millipore Sigma) was allowed to sit on the benchtop in a clear glass scintillation vial for one week. Initially, the compound was dark yellow and did not have any impurities visible by
1H NMR spectroscopy (Figure 12a). After one week, the compound was pale yellow and was observed to have decomposed by approximately 40% conversion, as monitored by
1H NMR spectroscopy (Figure 12b). In contrast, when a 10 mg sample (purchased from Millipore Sigma) was stored in a glass scintillation vial wrapped in aluminum foil for one week, no decomposition was observed. We hypothesize that this decomposition is due to a [2+2] photocycloaddition of
2-methylnaphthalene-1,4-dione (
3), resulting in a mixture of diastereomers. The resulting impurity cannot be removed from
3 through the workup described herein. As a result, we recommend that
3, particularly in the solid form, should be stored in the absence of light and exposure of the compound to light should be minimized during workup.
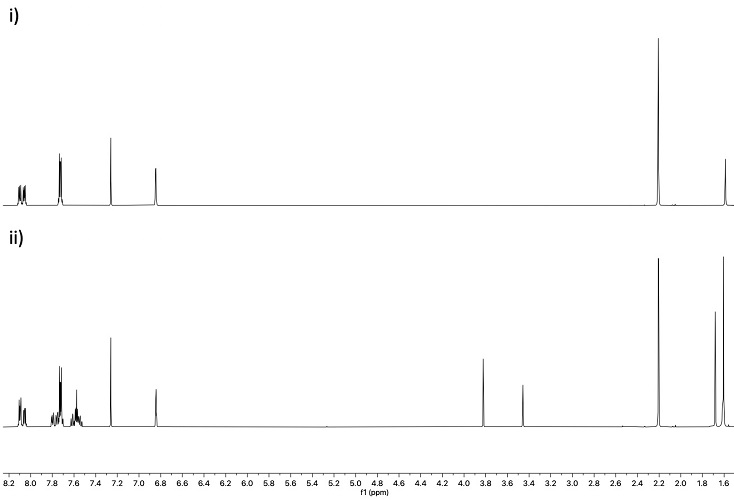
Figure 12. 1H NMR of 3 i) before and ii) after exposure to light in the solid form for one week (provided by checkers)
Working with Hazardous Chemicals
The procedures in
Organic Syntheses are intended for use only by persons with proper training in experimental organic chemistry. All hazardous materials should be handled using the standard procedures for work with chemicals described in references such as "Prudent Practices in the Laboratory" (The National Academies Press, Washington, D.C., 2011; the full text can be accessed free of charge at
http://www.nap.edu/catalog.php?record_id=12654. All chemical waste should be disposed of in accordance with local regulations. For general guidelines for the management of chemical waste, see Chapter 8 of Prudent Practices.
In some articles in Organic Syntheses, chemical-specific hazards are highlighted in red "Caution Notes" within a procedure. It is important to recognize that the absence of a caution note does not imply that no significant hazards are associated with the chemicals involved in that procedure. Prior to performing a reaction, a thorough risk assessment should be carried out that includes a review of the potential hazards associated with each chemical and experimental operation on the scale that is planned for the procedure. Guidelines for carrying out a risk assessment and for analyzing the hazards associated with chemicals can be found in Chapter 4 of Prudent Practices.
The procedures described in Organic Syntheses are provided as published and are conducted at one's own risk. Organic Syntheses, Inc., its Editors, and its Board of Directors do not warrant or guarantee the safety of individuals using these procedures and hereby disclaim any liability for any injuries or damages claimed to have resulted from or related in any way to the procedures herein.
3. Discussion
Ring forming strategies for
para-hydro- and
para-benzoquinone synthesis include the Wulff-Dötz reaction, Moore-Liebeskind rearrangement, Hauser-Kraus annulation, oxidative Bergman cyclization, and variants of the Diels-Alder reaction utilizing oxygenated furans as dienes (Figure 13A).
3 With respect to Diels-Alder approaches, most methodologies can be conceptualized as the application of a ketene equivalent as diene. For example, 1,4-tautomerization of
para-hydroquinone
4 reveals a Diels-Alder retron in dienone
6, which can be retrosynthetically disconnected back to oxy-substituted α,β-unsaturated ketene
6 as diene, and acetylene (
7) as dienophile (
Figure 13B). Generally speaking, ketenes are unsuitable reactants within the Diels-Alder reaction,
4 necessitating the development of synthetic equivalents for application in such contexts. The most common realization of this strategy proceeds via the coupling of 2-oxy-substituted furans
8 with acetylenic dienophiles, which following ring-opening and proton transfer yield
para-hydroquinones of general structure
4.
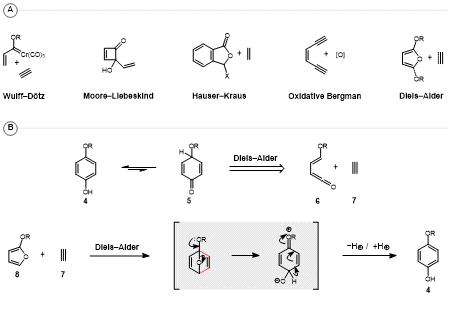
Figure 13. (A) Overview of annulative approaches toward para-quinones; (B) Prototypical Diels-Alder approach to para-hydroquinones
In 1980, Brownbridge and Chan disclosed the first synthesis of 2,5-bis(trimethylsilyloxy)furans
9 while also demonstrating their Diels-Alder reactivity with ethyl acrylate (
10) and dimethyl maleate (
11) (Figure 14A).
5 In analogy to the ring-opening chemistry of 2-oxy-substituted furans, treatment of the cycloadducts with aqueous NaF provided the corresponding
para-hydroquinones
12 or
13 in up to 98% yield. Alternatively, coupling the same furans with dimethyl acetylenedicarboxylate (
14) yielded
para-benzoquinones
15; however, competitive
para-hydroquinone formation was observed in most cases (Figure 14B). Conceptually, these furans can be considered as vicinal bisketene equivalents for application as dienes in the Diels-Alder reaction.
6Figure 14. Reactivity of 2,5-bis(trimethylsilyloxy)furans (9) with: (A) ethyl acrylate (10), dimethyl maleate (11), and (B) dimethyl acetylenedicarboxylate (14)
Within Chan's seminal report the 2,5-bis(trimethylsilyloxy)furan motif was described as "
very sensitive to moisture and air." Consequently, over the intervening 40 years these substrates have only been utilized in synthesis on a handful of occasions,
7 and reports detailing the preparation of new derivatives are scant.
8 In the interests of developing this chemistry into a more broadly applicable retrosynthetic disconnection, we recently demonstrated that bulkier
tert-butyldimethylsilyloxy variants of their trimethylsilyloxy congeners provide a significant improvement in furan stability.
6 As exemplified in Figure 15, ((3-methylfuran-2,5-diyl)bis(oxy))bis(
tert-butyldimethylsilane) (
1) exhibits an enhanced stability profile toward water, methanol, oxygen, heat, and buffered silica (pH = 7), facilitating multigram-scale preparation, purification, and long-term storage.
9,10,11,12,13,14,15,16,17Figure 15. Stability profile of 2,5-bis(silyloxy)furans 1 and 16
Representative para-hydroquinones synthesized from 2,5-bis(tert-butyldimethylsilyloxy)furans include indole 17, silyl enol ether 18, and BiPhePhos-inspired derivative 19 (Figure 16). Reaction with acetylenic dienophiles yield the corresponding para-benzoquinones (with no evidence for para-hydroquinone formation), and illustrative examples prepared by reaction with arynes as dienophiles include estrone-derived 20, and methoxy-substituted 21 (structurally pertinent daunorubicin aglycone pictured). The 2,5-bis(tert-butyldimethylsilyloxy) motif was also extended to pyrroles, enabling access to para-iminoquinone 22, or following reduction, the medicinally relevant para-aminophenol motif 23 (c.f. minocycline).
Figure 16. Selected substrate scope
2,5-Bis(tert-butyldimethylsilyloxy)furans, and related pyrroles, are readily accessible via the procedure described herein. They have been established as valuable building blocks for the convergent synthesis of highly substituted aromatics, including para-hydroquinones, para-benzoquinones, 1,4-napthoquinones, and para-iminoquinones. This modular protocol enables gram-scale access to these compounds, facilitating their application across various settings.
Appendix
Chemical Abstracts Nomenclature (Registry Number)
Methylsuccinic anhydride; (4100-80-5)
Triethylamine; (121-44-8)
tert-Butyldimethylsilyl trifluoromethanesulfonate; (69739-34-0)
2-Iodophenol; (533-58-4)
Pyridine; (110-86-1)
Trifluoromethanesulfonic anhydride; (358-23-6)
n-Butyllithium; (109-72-8)

|
Jessica Budwitz received her BS in Chemistry in May 2022 from the University of Georgia where she worked in the Newton group studying the application of Diels-Alder strategies in total synthesis. As of Fall 2022, Jessica is pursuing her Ph.D. within the Newton group. |

|
Christopher Newton studied chemistry at Victoria University of Wellington, New Zealand, received his Ph.D. with Michael Sherburn at the Australian National University, and did post-doctoral research with Nicolai Cramer at EPFL, Switzerland. He started his independent career as a DECRA Fellow at the University of Adelaide, Australia before relocating to the University of Georgia, USA. |

|
David J. Charboneau received his BS from Villanova University in 2015. Subsequently, he completed his Ph.D. at Yale University in 2021, where he performed research in the laboratory of Prof. Nilay Hazari developing cross-electrophile coupling reactions that employ homogeneous organic reductants. David then pursued postdoctoral research in the laboratory of Prof. Sarah Reisman, where his projects involved natural product synthesis and the discovery of samarium-catalyzed transformations. In early 2023, David will begin a position at Vertex Pharmaceuticals as a medicinal chemist. |

|
Jordan Thompson is a third-year graduate student in Professor Sarah Reisman's laboratory. He is interested in the total synthesis of complex natural products and completed his undergraduate studies at UC Berkeley under the guidance of Professor Tom Maimone. |
Copyright © 1921-, Organic Syntheses, Inc. All Rights Reserved