Org. Synth. 2003, 80, 18
DOI: 10.15227/orgsyn.080.0018
ASYMMETRIC SYNTHESIS OF N-tert-BUTOXYCARBONYL α-AMINO ACIDS. SYNTHESIS OF (5S,6R)-4-tert-BUTOXYCARBONYL-5,6-DIPHENYLMORPHOLIN-2-ONE
[(4-Morpholinecarboxylic acid, 6-oxo-2,3-diphenyl-, 1,1-dimethylethyl ester, (2S,3R)-)]
Submitted by Robert M. Williams, Peter J. Sinclair, Duane E. DeMong, Daimo Chen, and Dongguan Zhai
1.
Checked by Wenlin Lee and Marvin J. Miller.
1. Procedure
A. Ethyl (1'S,2'R)-N-(1',2'-diphenyl-2'-hydroxyethyl)glycinate. A 1-L. three-necked, round-bottomed flask, equipped with a magnetic stirring bar, pressure-equalizing addition funnel, and two stoppers, is charged with 25 g (0.117 mol) of (1S,2R)-1,2-diphenyl-2-hydroxyethylamine (Note 1), 29 g (0.176 mol) of ethyl bromoacetate and 455 mL of anhydrous tetrahydrofuran (THF) (Note 2). To the stirred mixture, 24 g (0.234 mol) of triethylamine (Note 3) is added dropwise via the addition funnel. After stirring vigorously for 20 hr at room temperature, the solids are removed by filtration and washed three times with THF (Note 4). The clear yellowish filtrate and washes are concentrated on a rotary evaporator (to remove solvent, excess triethylamine, and ethyl bromoacetate) to afford an off-white solid, which is collected with a Buchner funnel, then washed with water (Note 5) to dissolve residual salts. The residue is air-dried in the filter funnel with water aspirator vacuum. The crude product is dissolved in 117 mL of boiling absolute ethanol (EtOH) and stored at room temperature overnight. White crystals are collected by filtration, washed with ice-cold (0°C) absolute EtOH carefully, and dried at 50°C (Note 6). The above described procedure is done in duplicate, providing ethyl (1'S,2'R)-N-(1',2'-diphenyl-2'-hydroxyethyl)glycinate in yields of 29.01 g and 28.96 g, (average yield: 83%) (Notes 7, 8).
B.
Ethyl (1'S,2'R)-N-tert-butyloxycarbonyl-N-(1',2'-diphenyl-2'-hydroxyethyl-glycinate.
3 A
1-L, three-necked, round-bottomed flask, equipped with a
magnetic stirring bar,
reflux condenser, and two stoppers, is charged with
167 mL of a solution of saturated aqueous sodium bicarbonate (NaHCO
3),
34.2 g (0.585 mol) of sodium chloride (NaCl),
350 mL of chloroform (CHCl
3)
(Note 9),
25 g (0.084 mol) of ethyl (1'S,2'R)-N-(1',2'-diphenyl-2'-hydroxyethyl)glycinate, and
36.5 g (0.167 mol) of di-tert-butyl dicarbonate (Note 10). The resulting mixture is then heated to reflux for 20 hr and vigorously stirred.
The reaction mixture is placed in a separatory funnel; the aqueous phase is separated and extracted twice with chloroform (CHCl3). The organic phases are combined, washed twice with water, and dried with anhydrous sodium sulfate overnight. After filtration to remove sodium sulfate, concentration under vacuum affords a residue, which is heated (5 mm, 130°C oil bath) to remove and recover excess di-tert-butyl dicarbonate (Note 11). A total of 33.2 g (83 mmol) of crude ethyl (1'S,2'R)-N-tert-butyloxycarbonyl-N-(1',2'-diphenyl-2'-hydroxyethyl)glycinate, is obtained as an oil and used directly in the subsequent lactonization reaction.
C. (5S,6R)-4-tert-Butoxycarbonyl-5,6-diphenylmorpholin-2-one. A 2-L, round-bottomed flask, equipped with a magnetic stirring bar, is charged with 33.2 g (0.083 mol) of crude ethyl (1'S,2'R)-N-tert-butyloxycarbonyl-N-(1',2'-diphenyl-2'-hydroxyethyl) glycinate, 1.90 g (0.010 mol) of p-toluenesulfonic acid monohydrate (Note 12), and 670 mL of benzene (Notes 13, 14). The reaction flask is fitted with a Soxhlet extractor, which is packed with 65 g of calcium chloride (CaCl2, anhydrous, 30 mesh) and a condenser with a drying tube. The reaction mixture is heated to reflux with stirring for 10 hr (Note 15). After the first 4 hr, the CaCl2 in the Soxhlet extractor is replaced; the total amount of CaCl2 used in the reaction is 130 g. Ultimately, the reaction mixture becomes a suspension. Removal of benzene under reduced pressure (Note 16) yields white solids that are dissolved in 310 mL of dichloromethane (CH2Cl2). The solution is washed with water and 5% aqueous NaHCO3 in a separatory funnel until the CH2Cl2 solution becomes clear. After removal of CH2Cl2 under vacuum, the resulting white crystals are dissolved by boiling in 600 mL of absolute EtOH; the resulting solution is allowed to stand at room temperature overnight. The white crystals are collected by filtration, washed with ice-cold EtOH, and dried at 60°C. Concentration of the mother liquor by rotary evaporation affords a residue that can also be recrystallized in absolute EtOH to provide additional product. The above described procedure is performed in duplicate, providing (5S,6R)-4-tert-butyloxycarbonyl-5,6-diphenylmorpholin-2-one in yields of 21.7 g and 20.3 g (average yield for the two steps: 71%) (Notes 17-19).
2. Notes
1.
(1S,2R)- and (1R,2S)-1,2-Diphenyl-2-hydroxyethylamine (>98% ee) were prepared according to the procedure of Tishler, et al.2 The checkers obtained the amino alcohols from Aldrich Chemical Co., Inc.2.
Tetrahydrofuran was dried by distillation from
sodium-benzophenone ketyl.
3.
Triethylamine was dried by distillation from CaH
2.
4.
Reagent grade or technical grade (i.e. undistilled) THF can be used for this wash.
5.
Extensive washing in a Buchner funnel with water is recommended.
6.
The product was dried in a
crystallizing dish in a
vacuum oven.
7.
The physical and spectroscopic properties for
ethyl (1'S,2'R)-N-(1',2'-hydroxyethyl)glycinate are as follows:
mp. 127-128°C,
[α]D25 +24.4 (c 5.5, CH2Cl2); IR (NaCl, CDCl
3) cm
−1: 3840-3430, 3330, 3080, 3045, 2995, 2940, 1750, 1460, 1385, 1210, 1035, 915, 740;
1H NMR (270 MHz, CDCl
3,) δ: 1.20 (3H, t, J = 7.1), 2.2 (2H, br s), 3.15 (1H, 1/2 AB q, J = 17.5), 3.29 (1H, 1/2 AB q, J = 17.5), 3.95 (1H, d, J = 6.0), 4.11 (2H, q, J = 7.1), 4.80 (1H, d, J = 6.0), 7.17-7.32 (10H, m); Anal. Calcd. for C
18H
21NO
3: C, 72.22; H, 7.07; N, 4.68. Found: C, 72.31; H, 7.16; N, 4.56.
8.
From the antipode
(1R,2S)-1,2-diphenyl-2-hydroxyethylamine is obtained the corresponding enantiomer
ethyl (1'R,2'S)-N-(1',2'-diphenyl-2'-hydroxyethyl)-glycinate.
mp 127-128°C,
[α]D25 −24.4 (c 5.5, CH2Cl2). This series, also performed in duplicate, provides yields of
29.2 g and
29.5 g (average yield:
84%).
9.
Technical grade chloroform was used.
10.
Di-tert-butyl dicarbonate was purchased from Aldrich Chemical Co., Inc.11.
The recovered
di-tert-butyl dicarbonate is very pure and can be reused.
12.
p-Toluenesulfonic acid monohydrate was purchased from Aldrich Chemical Co., Inc.13.
ACS grade benzene was purchased from Fisher Scientific.
14.
The checkers found that
770 mL of cyclohexane could also be used as the solvent for this step, avoiding the use of large amounts of toxic
benzene. The crude Step B product, a white solid, very slowly dissolves in refluxing
cyclohexane; upon conversion to the morpholinone, which is insoluble in
cyclohexane, the product precipitates out of solution. The reaction mixture remains a suspension throughout the reaction, but the texture of the solids shows a subtle change over time, indicating the progress of the reaction. The color of the solution also turns slightly yellow. The reaction time is extended to 17 hr and the CaCl
2 pellets in the Soxhlet extractor are changed after the first 5-6 hr. Work up of the reaction is identical to that described in the procedure.
15.
The Soxhlet extractor does not require the standard Soxhlet filter thimbles; cotton above and below the CaCl
2 pellets is sufficient.
16.
Caution! Care should be exercised in performing the reaction and concentrating benzene in a fume hood.
17.
The physical and spectroscopic properties for
(5S,6R)-4-tert-butyloxycarbonyl-5,6-diphenylmorpholin-2-one are as follows:
mp 207°C;
[α]D25 +86.3 (c 5.6, CH2Cl2); IR (NaCl, CH
2Cl
2) cm
−1: 3050, 2975, 1755, 1690, 1380, 1255, 1150, 1100, 1045;
1H NMR (200 MHz) (DMSO-d
6 vs. DMSO) (120°C) δ: 1.25 (9H, s), 4.52 (2H, d, J = 1.1), 5.16 (1H, d, J = 3.0), 6.17 (1H, d, J = 3.0), 6.63-6.68 (2H, m), 7.0-7.3 (8H, m); Anal. calcd. for C
21H
23NO
4: C, 71.37; H, 6.56; N, 3.96. Found: C, 71.08; H, 6.44; N, 3.98. The
checkers obtained
22.7 g, (
77%) after the first recrystallization;
mp 205-207°C; [α]D25 −86.0 (c 1.0, CH2Cl2).
18.
From the antipode,
ethyl (1'R,2'S)-N-(1',2'-diphenyl-2'-hydroxyethyl)-glycinate, is obtained the corresponding enantiomer,
(5R,6S)-4-tert-butyloxycarbonyl-5,6-diphenylmorpholin-2-one,
mp 207°C,
[α]D25 +85.8 (c 5.7, CH2Cl2). This series, also performed in duplicate, provides yields of
24.0 g and
23.6 g, respectively (average yield for the two steps:
80%).
19.
Optical rotations were measured on a Rudolf Research Autopol III automatic polarimeter operating at 589 nm, corresponding to the
sodium D line.
Handling and Disposal of Hazardous Chemicals
The procedures in this article are intended for use only by persons with prior training in experimental organic chemistry. All hazardous materials should be handled using the standard procedures for work with chemicals described in references such as "Prudent Practices in the Laboratory" (The National Academies Press, Washington, D.C., 2011 www.nap.edu). All chemical waste should be disposed of in accordance with local regulations. For general guidelines for the management of chemical waste, see Chapter 8 of Prudent Practices.
These procedures must be conducted at one's own risk. Organic Syntheses, Inc., its Editors, and its Board of Directors do not warrant or guarantee the safety of individuals using these procedures and hereby disclaim any liability for any injuries or damages claimed to have resulted from or related in any way to the procedures herein.
3. Discussion
(+)- and (−)-4-tert-Butoxycarbonyl-5,6-diphenylmorpholin-2-one can be used for the asymmetric synthesis of α-amino acids.
3 The methylene carbon of the morpholinone can be efficiently brominated with NBS in CCl
4 to give a single diastereomeric bromide (anti). This bromo lactone serves as a useful electrophilic glycine synthon that undergoes stereoselective coupling with a variety of organometallic reagents. In general, coupling proceeds with net retention of stereochemistry furnishing the crystalline anti-homologation products. Dissolving metal reduction (
lithium or
sodium in liquid NH
3/EtOH/THF) directly furnishes the corresponding N-tert-butoxycarbonyl-protected amino acid derivative. Alternatively, the N-t-Boc group can be removed (TFA or HCO
2H) and catalytic hydrogenation (PdCl
2/EtOH/THF, 25°C, 40 psi) furnishes the free zwitterionic amino acid. In some cases, coupling has been observed to proceed with net inversion of stereochemistry to furnish syn-homologation products as oils. In cases where modest stereoselectivity is observed, the syn- and anti-isomers can be readily separated by chromatography. If the coupling is anti-selective (which is typical), a simple recrystallization will consistently give the final amino acid in >98% ee.
The lactones are quite stable to storage and handling (shelf life at room temperature >16 years). Treatment of these substances with either lithium, sodium or potassium hexamethyldisilazane in THF at −78°C generates the corresponding enolate that can be stereoselectively alkylated with an appropriate electrophile to give anti-homologated products. Similar reductive processing of these materials provides amino acid derivatives in high enantiomeric excess.
The present method offers a versatile and practical protocol for preparing either D- or L- amino acids and offers the unique advantage of allowing direct access to the N-t-Boc derivatives. Conversion of the diphenyl amino alcohol moiety into the desired product and the water-insoluble bibenzyl, which is removed by simple extraction into ether or pentane, by either reductive protocol (1 and 2, shown below) is noteworthy. The water-soluble zwitterions, or in the case of the N-t-Boc amino acids, the N-t-Boc carboxylate salts, remain in the aqueous phase and are conveniently isolated in high chemical purity without, in most cases, the need for additional time-consuming and expensive chromatographic purification steps. With the oxidative methods (3 and 4), the diphenyl amino alcohol moiety is converted into two equivalents of benzaldehyde which can generally be separated from the amino acid by extraction. The ease and convenience of processing the derivatized morpolinones to the desired amino acids is testament to the flexibility and reliability of this methodology.
|
|
Method
|
Conditions
|
Type of 'R' Group
|
Amino Acid
|
|
|
1) Birch Reduction
|
(Li or Na, NH3(l))
|
Unsaturated; Saturated, Aliphatic
|
N-t-Boc
|
|
2) Catalytic hydrogenation (after removal of the Boc group)
|
(H2 / Pd-C, EtOH)
|
Saturated, Aliphatic
|
Zwitterion
|
|
3) Oxidation
|
1) H+; 2) NaIO4
|
Aromatic; Saturated, Aliphatic
|
Zwitterion
|
|
4) Oxidation
|
1) H+; 2) Pb(OAc)4
|
Aromatic β-Arylcyclopropyl Saturated, Aliphatic
|
Methyl ester
|
|
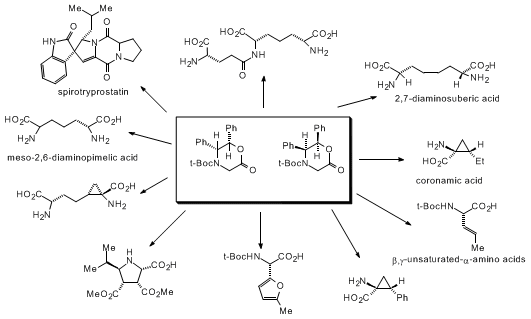
As illustrated in the accompanying procedure, (N-t-Boc)allylglycine, this morpholinone has proven to be one of the most versatile and useful chiral glycine equivalents from which a large variety of amino acids can be prepared. Several other, recent asymmetric electrophilic glycine enolate and cation equivalents have been devised.
4As shown in the scheme above, no other chiral glycine equivalent (from the laboratories of Seebach, Oppolzer, Evans, Myers, Schöllkopf, etc.) can provide the range of reactivity of this substance.
The chemistry of these compounds was reviewed in 1992
3j and again in 1995
3q and there are numerous publications and patent citations in the literature from other laboratories that have employed these synthons; a few examples are cited.
5,6,7,8,9,10,11,12,13,14,15,16,17 Finally, the N-t-Boc-protected morpholinones have been used extensively in our laboratory to prepare a variety of amino acids and peptide isosteres as shown below.
3 These examples serve to illustrate the great versatility that these glycine derivatives offer for preparing structurally diverse amino acids in high optical purity.
Appendix
Chemical Abstracts Nomenclature (Collective Index Number);
(Registry Number)
Ethyl bromoacetate:
Acetic acid, bromo-, ethyl ester (8,9); (105-36-2)
Triethylamine:
N,N-Diethylethanamine (9); (121-44-8)
Di-tert-butyl dicarbonate:
Dicarbonic acid, bis(1,1-dimethylethyl) ester (9); (24424-99-5)
p-Toluenesulfonic acid:
Benzenesulfonic acid, 4-methyl-, monohydrate (9); (6192-52-5)
(1S,2R)-1,2-Diphenyl-2-hydroxyethylamine:
Benzeneethanol, β-amino-α-phenyl-, (αR,βS)-rel- (9); (23412-95-5)
(1R,2S)-1,2-Diphenyl-2-hydroxyethylamine:
Benzeneethanol, β-amino-α-phenyl-, (αS,βR)- (9); (23364-44-5)
Ethyl (1'S,2'R)-N-(1',2'-diphenyl-2'-hydroxyethyl)glycinate:
Glycine, N-(2-hydroxy- 1,2-diphenylethyl)-, ethyl ester, [R-(R*,S*)]- (9); (100678-82-8)
Ethyl (1'R,2'S)-N-(1',2'-diphenyl-2'-hydroxyethyl)glycinate:
Glycine, N-(2-hydroxy- 1,2-diphenylethyl)-, ethyl ester, [S-(R*,S*)]- (9); (112835-62-8)
(1'S,2'R)-Ethyl N-tert-butyloxycarbonyl-N-(1',2'-diphenyl-2'-hydroxyethyl)glycinate:
Glycine, N-[(1,1-dimethylethoxy)carbonyl]-N-(2-hydroxy-1,2-diphenylethyl)-, ethyl ester, [R-(R*,S*)]- (9); (112741-70-5)
(1'R,2'S)-Ethyl N-tert-butyloxycarbonyl-N-(1',2'-diphenyl-2'-hydroxyethyl)glycinate:
Glycine, N-[(1,1-dimethylethoxy)carbonyl]-N-(2-hydroxy-1,2-diphenylethyl)-, ethyl ester, [S-(R*,S*)]- (9); (112741-73-8)
(5S,6R)-4-tert-Butyloxycarbonyl-5,6-diphenylmorpholin-2-one:
4-Morpholinecarboxylic acid, 6-oxo-2,3-diphenyl-, 1,1-dimethylethyl ester, (2R-cis)- (9); (173397-90-5)
(5R,6S)-4-tert-Butyloxycarbonyl-5,6-diphenylmorpholin-2-one:
4-Morpholinecarboxylic acid, 6-oxo-2,3-diphenyl-, 1,1-dimethylethyl ester, (2S-cis)- (9); (112741-50-1)
Copyright © 1921-, Organic Syntheses, Inc. All Rights Reserved