Org. Synth. 2003, 80, 144
DOI: 10.15227/orgsyn.080.0144
SYNTHESIS AND [3+2] CYCLOADDITION OF A 1,1-DIALKOXY-2-METHYLENECYCLOPROPANE: 6,6-DIMETHYL-1-METHYLENE-4,8-DIOXASPIRO[2.5]OCTANE and cis-5-(5,5-DIMETHYL-1,3-DIOXAN-2-YLIDENE)HEXAHYDRO-1(2H)-PENTALEN-2-ONE
[(4,8-Dioxaspiro[2.5]octane, 6,6-dimethyl-1-methylene-)]
Submitted by Masaharu Nakamura
1, Xiao Qun Wang
1, Masahiko Isaka
1, Shigeru Yamago
2, and Eiichi Nakamura
1.
Checked by Jimmy Wu and Edward J. J. Grabowski.
1. Procedure
Caution! All operations should be performed in a well-ventilated hood, and care should be taken to avoid skin contact with 1,3-dichloro-2-propanone and cyclopropenes.
A. 2,2-Bis-(chloromethyl)-5,5-dimethyl-1,3-dioxane (1). A mixture of 1,3-dichloroacetone (152 g, 1.20 mol, an irritant), neopentyl glycol (138 g, 1.32 mol), p-toluenesulfonic acid (4.6 g, 0.024 mol) and benzene (100 mL) (Note 1) is heated to reflux for 19 hr in a 500-mL, round-bottomed flask equipped with a Dean-Stark trap and a condenser with azeotropic removal of water. After separation of water ceases, the reaction mixture is partitioned between hexane (500 mL) and saturated sodium bicarbonate (NaHCO3) (200 mL). The organic phase is washed with water (100 mL) and saturated sodium chloride (NaCl) (100 mL), dried over magnesium chloride (MgSO4), and concentrated on a rotary evaporator. Distillation of the residue yields, after about 5 g of forerun, 249 g (97%) of the acetal 1 as a colorless oil (bp 99-100°C, 3.5 mm) (Note 2).
B. 1,6,6-Trimethyl-4,8-dioxaspiro[2.5]oct-1-ene (3) (Note 3). A solution of sodium amide in liquid ammonia is prepared according to a procedure previously described (Notes 4, 5). An oven-dried, 2-L, three-necked, round-bottomed flask is equipped with a mechanical stirrer, nitrogen gas inlet, and dry ice/acetone condenser protected with a drying tube containing potassium hydroxide pellets. The flask is flushed with nitrogen introduced through the gas inlet, then is placed in a dry ice/acetone bath. The nitrogen source is replaced with a hose connected to a cylinder of ammonia (NH3). Gaseous NH3 is introduced to the flask condensing ca. 400 mL of NH3 and gentle stirring is started. The NH3 inlet is replaced with a glass stopper and the dry ice/acetone bath is replaced with a −35°C bath (electronically controlled or a dry ice/trichloroethylene bath). Crystals of Fe(NO3)3·9H2O (0.3 g) are added through the neck having a stopper. To the resulting orange solution is added a small (about 5 mm3) cube of sodium. The solution is stirred (Note 6) until the blue color disappears and fine black particles appear. Pieces of sodium (21.44 g, 0.930 mol) are then added over 35 min. After 20 min, the solution turns into a dark gray suspension with a white precipitate. The cooling bath is replaced with a dry ice/acetone bath, and the gas inlet is replaced with a pressure-equalizing addition funnel containing a solution of 2,2-bis-(chloromethyl)-5,5-dimethyl-1,3-dioxane 1 (63.93 g, 0.300 mol) in 150 mL of dry ether (Et2O). This solution is added dropwise to the slurry of sodium amide in liquid NH3 over 1 hr. The addition funnel is rinsed with 20 mL of dry Et2O. The cooling bath is removed, and the mixture is stirred for 3 hr (Note 7), then is cooled again with a dry ice/acetone bath. After 10 min, a solution of freshly distilled iodomethane (44.71 g, 0.315 mol) in 80 mL of dry Et2O is added via the addition funnel over 1 hr (Notes 8, 9); the funnel is rinsed with 20 mL of dry Et2O. After stirring for 15 min, the cooling bath is removed and the mixture is stirred for 1 hr. The mixture is cooled with a dry ice/acetone bath again and solid ammonium chloride (NH4Cl) (20.24 g, 0.378 mol) is added in several portions over 5 min. The dry ice condenser is removed and the ammonia is allowed to evaporate. The cooling bath is replaced with a water bath (ca. 30°C), and a 1:1 mixture of dry Et2O and dry pentane (400 mL) is added through the addition funnel over 10 min with vigorous stirring. The water bath temperature is maintained between 25-30°C. After evaporation of most of the ammonia (1.5 to 2 hr), the ethereal solution is filtered by suction through a pad of Hyflo Super Cel to remove the inorganic salts. The filter cake is washed three times with 80 mL of Et2O. The combined filtrate and washes are concentrated under reduced pressure (30-40 mm, 25°C) (Note 10) and the residue is distilled to yield 3 as a colorless oil (32.3 g, 70%; bp 58-61°C, 6-7 mm) (Notes 11-14). While this material is adequate for use in Step C, a pure sample can be obtained by flash chromatography of the crude product followed by distillation. For 36.1 g of crude product (obtained on a 0.25 mol scale), chromatography is performed with 290 g of silica gel (Merck, Kieselgel 60) and Et2O:pentane (5:95) as eluent, collecting 160-mL fractions. Fractions 3-16 containing the product are combined. Removal of the solvent under reduced pressure followed by vacuum distillation afforded 29.3 g of 3 in 76% yield (99% pure by GC).
C. 6,6-Dimethyl-1-methylene-4,8-dioxaspiro[2.5]octane (5). An oven-dried, 100-mL, round-bottomed flask, equipped with a magnetic stirring bar and three-way stopcock, which is connected to a vacuum/nitrogen line, is flushed with nitrogen. The flask is placed in a plastic bag filled with nitrogen, and powdered potassium tert-butoxide (1.32 g, 11.8 mmol) (Note 15) is introduced quickly. The flask is connected to a nitrogen source, tert-butyl alcohol (7.11 g, 96 mmol) and 40 mL of Et2O (Note 16) are introduced by syringe, and the solution is stirred for 10 min at room temperature.
An oven-dried 300-mL, round-bottomed flask, equipped with a magnetic stirring bar and three-way stopcock, connected to a vacuum/nitrogen line, is charged with 1,6,6-trimethyl-4,8-dioxaspiro[2.5]oct-1-ene (3) (46.3 g, 0.30 mol) under nitrogen. Ether (100 mL) is introduced via syringe, and the solution is stirred with external cooling at −78°C (dry ice/acetone bath). The solution of potassium tert-butoxide is added to this solution through a 1.5 mm i.d. cannula over 5 min. After stirring for 5 min at this temperature, the cooling bath is removed and the solution is allowed to warm to room temperature (Note 17). After stirring for 4 hr (Note 18), 15 mL of 1N HCl is added in one portion. The mixture is transferred to a separatory funnel with the aid of 30 mL of Et2O, and the aqueous phase is separated. The organic phase is washed successively with 5 mL each of saturated NaHCO3 and saturated NaCl, and is dried over Na2SO4. After concentration on a rotary evaporator, a pale yellow oil is obtained. Distillation under reduced pressure (Note 19) (57-59°C,7.3 mm) affords 36.49 g (79% yield) of 5 (Notes 19, 20) after ca. 1.5 g of a forerun. The material is sufficiently pure for the subsequent step.
D. cis-5-(5,5-Dimethyl-1,3-dioxan-2-ylidene)hexahydro-1(2H)-pentalen-2-one (6). An oven-dried, 50-mL, round-bottomed flask, equipped with a magnetic stirring bar and three-way stopcock, is flushed with nitrogen. A mixture of 6,6-dimethyl-1-methylene-4,8-dioxaspiro[2.5]octane, 5, (8.48 g, 55 mmol) and 2-cyclopenten-1-one (4.52 g, 55 mmol) in 15 mL of acetonitrile is introduced via syringe and the solution is heated at 60°C for 12 hr. The three-way stopcock is replaced with a distillation head. Solvent is removed by distillation (ca. 30-120°C/ca. 20-1.4 mm) and the residue is distilled under reduced pressure (142-143°C, 1.4 mm) to afford 6 (10.0 g, 42.3 mmol, 77%), which crystallizes upon standing at room temperature (Notes 21, 22).
2. Notes
1.
The following chemicals were purchased from Kanto Chemical Co. Inc. and used as received: neopentyl glycol, p-toluenesulfonic acid, sodium, and n-BuLi.
N,N,N',N'-Tetramethylethylenediamine was purchased from Aldrich Chemical Company, Inc. and distilled before use.
1,3-dichloro-2-propanone was obtained from Wacker Chemicals East Asia and used as received.
Reagent grade benzene, pentane, ether, THF, tert-butyl alcohol, acetonitrile, and toluene were purchased from Wako Chemicals Industries Ltd. Benzene,
pentane, and
tert-butyl alcohol were distilled from CaH
2;
ether and THF from
sodium benzophenone ketyl immediately before use;
acetonitrile successively from P
2O
5 and anhydrous K
2CO
3.
Potassium tert-butoxide and 2-cyclopenten-1-one were purchased from Tokyo Kasei Kogyo Co. Ltd.; the ketone was distilled before use.
2.
The product was pure by 270 MHz
1H NMR analysis. Spectral properties: IR (neat) cm
−1: 2950, 2860, 1105, 1025, 770;
1H NMR (270 MHz, CDCl
3) δ: 1.00 (s, 6 H), 3.57 (s, 4 H), 3.80 (s, 4 H).
3.
Important notes: (1) Since the reactions are sensitive to temperature effects, care must be taken to allow the reaction mixture to reach thermal equilibrium with the cooling bath at each stage of the operation. (2) Because of the rather unstable nature of the product in its impure form, the entire procedure except the final distillation must be carried out within a day. This will take at least 12 hr. See also
(Note 10).
4.
One of the procedures for converting
sodium to sodium amide described in
Organic Syntheses is used.
35.
The use of commercial NaNH
2 is not recommended since the yield and the purity of the cyclization product are heavily dependent on the purity of NaNH
2.
6.
Gentle stirring (e.g., 120 rpm) is recommended to avoid loss of the amide base which may adhere to the upper part of the flask. This was found to be a good practice to achieve the most accurate base/substrate molar ratio.
7.
The reaction may be complete in about 2 hr. An aliquot may be removed and analyzed by GC for the intermediate
2-chlorocyclopropanone acetal. In case of a deficiency of sodium amide, a small amount of the chloride may remain but can be removed by distillation. The analysis may be performed on a 0.25 mm i.d. × 25 m capillary GC column (HR-1, Shinwa Chemical Industries, Ltd.) at 80°C. Typical retention times for the chlorocyclopropanone acetal and
3 are 9.9 and 4.6 min, respectively. The amount of the former may be estimated by comparison of its peak area with that of
3 assuming equal detector response factors of the two compounds.
8.
Rapid addition of
iodomethane causes the formation of a significant amount of 2,3-dialkylated compound and
2-methylenecyclopropanone acetal (by isomerization of the olefin).
9.
If
iodomethane is replaced with solid NH
4Cl (3≈4 eq to
1, added very carefully in several portions), the unsubstituted
cyclopropenone acetal 4 can be obtained in the same manner. The submitters carried out this reaction on a 0.3-1 mol scale in
70-85% distilled yield.
10.
Care must be taken not to lose material upon concentration. In case of a lack of time, the crude product may be kept under
nitrogen in a freezer (−20°C) and distilled later.
11.
In order to obtain a good yield, it is important to carry out the distillation as fast as possible (<25 min), but with the bath temperature below 100°C.
12.
This product may contain 6≈8% of the unsubstituted
cyclopropenone acetal 4 and 3≈5% of
2,3-dimethylcyclopropenone acetal and ca. 1% of
methylene-cyclopropanone acetal 5 as determined by capillary GC analysis on a 0.25 mm i.d. × 25 m capillary column (HR-1) at 80°C. Typical retention times for
3,
4,
5, and 2,3-dimethyl cyclopropenone acetal are 6.4, 4.3, 5.8 and 9.6 min, respectively. The checkers used material of this purity for Step C.
13.
The product has the following physical properties: IR (neat) cm
−1: 2950, 1840 (w), 1735, 1280, 1070;
1H NMR (270 MHz, CDCl
3) δ: 0.99 (s, 3 H, CH
3), 1.07 (s, 3 H, CH
3), 2.19 (3 H, cyclopropenyl CH
3), 3.63 (s, 4 H, OCH
2C), 7.34 (s, 1 H, C=CH);
13C NMR (100 MHz, CDCl
3) δ: 10.27, 22.14, 22.45, 30.47, 77.10, (2C), 83.24, 116.04, 125.42.
14.
The cyclization in Step B is an improvement of Butler's procedure for the synthesis of
4,
4,5 which employs less convenient reagents, KNH
2 and
1-bromo-3-chloroacetone acetal. Beside the acetals derived from
neopentyl glycol, those derived from
ethanol,
1,3-propanediol and
2,4-pentanediol have been synthesized by the present method.
8a The second part of Step B involves the formation and the electrophilic trapping of cyclopropenyl anion
2, which is the key element of the present preparations. Step B provides a simple route to substituted cyclopropenones, but the reaction is limited to alkylation with alkyl halides. The use of lithiated and zincated cyclopropenone acetal,
8 on the other hand, is more general and permits the reaction with a variety of electrophiles; alkyl, aryl and vinyl halides, Me
3SiCl, Bu
3SnCl, aldehydes, ketones, and epoxides. Repetition of the lithiation/alkylation sequence provides disubstituted cyclopropenone acetals.
15.
High purity of
potassium tert-butoxide is crucial for a clean reaction. The
tert-butanol must be anhydrous.
16.
A 1:1 mixture of dry
dimethylsulfoxide/Et
2O was successfully used for other higher homologues of alkylidenecyclopropanone acetals.
17.
The reaction can be monitored either by TLC or GC. TLC analysis may be performed with a plate (Merck No. 5765), eluting with a 1:9 mixture of
ethyl acetate and
hexane. The R
f values of
3 and
5 are 0.22 and 0.44, respectively. GC analysis performed under the same conditions as described in
(Note 12) separates these two compounds, which have retention times of 6.4 and 5.8 min, respectively.
18.
In the event that the reaction does not start or has not been completed, another 0.6-0.9 g portion of
potassium tert-butoxide dissolved in t-BuOH/Et
2O may be added.
19.
It is important to keep the bath temperature as low as possible. Distillation above 100°C would result in greatly reduced yield due to thermal decomposition of
5.
20.
The product has the following physical properties: IR (neat) cm
−1: 3150 (w), 1270, 1255, 1245, 1150, 1140, 1070, 1030, 1015, 970, 905;
1H NMR (200 MHz, CDCl
3) δ: 1.04 (s, 3 H, CH
3), 1.06 (s, 3 H, CH
3), 1.62 (dd, 2 H, J = 3.1, 2.4, cyclopropyl CH
2), 3.63 (s, 4 H, OCH
2C), 5.45 (t, 1 H, J = 2.4, C = CHH), 5.81 (t, 1 H, J = 3.1, C = CHH); Anal. Calcd for C
9H
14O
2: C, 70.10; H, 9.15. Found: C, 70.02; H, 9.25.
21.
The cycloadduct has the following spectral properties:
1H NMR (500 MHz, CD
3CN) δ: 0.96 (s, 3 H), 0.99 (s, 3 H), 1.64-1.70 (m, 1 H), 2.00 (br dd, 1 H, J = 15.6, 4.6), 2.02-2.10 (m, 1 H), 2.21 (distorted t, 2 H, J = 7.6), 2.33 (br dd, 1 H, J = 16.5, 3.7), 2.39-2.49 (m, 2 H), 2.52 (ddd, 1 H, J = 9.6, 4.7, 2.1) 2.74-2.81 (m, 1 H), 3.59 (d, 1 H, J = 7.3), 3.60 (d, 1 H, J = 7.3), 3.61 (s, 2 H);
13C NMR (125 MHz, CD
3CN) δ: 22.13 (q), 22.18 (q), 26.57 (t), 29.80 (t), 31.26 (s), 33.19 (t), 37.46 (t), 42.21 (d), 53.25 (d), 77.42 (t), 77.44 (t), 97.92 (s), 148.69 (s), 222.44 (s).
The ketene acetal functionality of the cycloadduct is moisture sensitive. It can be readily hydrolyzed to the
cyclopentanone ester 7 upon treatment with aqueous
acetic acid. Spectral properties of
7 are as follows: the major, more polar product-IR (neat) cm
−1: 3440, 1730;
1H NMR (200 MHz, CDCl
3) 0.91 (s, 6 H), 1.60 (dt, 1 H, J = 13.3, 9.7), 1.74-2.49 (m, 8 H), 2.61 (dt, 1 H, J = 9.5, 5.9), 2.71-2.99 (m, 2 H), 3.29 (br s, 2 H), 3.92 (s, 2 H); Anal. Calcd for C
14H
22O
4: C, 66.11; H, 8.72. Found: C, 66.00; H, 8.72; the minor, less polar product-IR (neat) cm
−1: 3440, 1730, 1720;
1H NMR (200 MHz, CDCl
3) δ: 0.92 (s, 6 H), 1.49-1.86 (m, 2 H), 2.03-2.37 (m, 7 H), 2.57-2.84 (m, 2 H), 2.85-3.06 (m, 1 H), 3.29 (br d, 2 H, J = 2.3), 3.94 (s, 2 H); Anal. Calcd for C
14H
22O
4: C, 66.11; H, 8.72. Found: C, 66.00; H, 8.86.
22.
This product may contain ca. 5% of impurities as judged by capillary GC analysis on a 0.25 mm i.d. × 25 m capillary column (HR-1) at 170°C. Typical retention times for the title compound and the side product are 8.8 and 4.5 min respectively.
3. Discussion
The procedures described herein illustrate the preparation of a substituted cyclopropenone acetal and an alkylidene cyclopropanone acetal.
7 The latter compound has been used to generate a dipolar trimethylenemethane (TMM) species that undergoes [3+2] cycloaddition with electron-deficient 2p-electron C=C and C=X compounds.
7The substituted cyclopropenone acetal synthesized in Step B can be readily hydrolyzed to the corresponding cyclopropenone.
8 This synthetic sequence provides the best and most versatile current synthetic route to substituted cyclopropenones.
9 The deficiencies of conventional procedures are precisely the synthesis of cyclopropenones with aliphatic substituents and functional groups, for which the present method has proven to be particularly useful.
10Cyclopropenones show considerable biological activity,
11,12 and have recently been employed as a key structural unit for a novel inhibitor of a cysteine protease.
13 The utility of cyclopropenone acetals has recently been recognized for vinylcarbene formation,
14,15 asymmetric synthesis,
16 and other processes.
17 Cycloaddition reactions of cyclopropenone acetals and congeners have also proven to be useful for chiral functionalization of buckminsterfullerenes.
18Step C describes a synthesis of the 1,1-dialkoxy-2-methylenecyclopropane (
5) from 1,1-dialkoxy-2-methylenecyclopropene (
3).
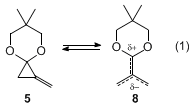
19 As described in Step D, upon thermolysis under mild conditions,
5 undergoes a [3+2] cycloaddition reaction by the reversible generation of a reactive dipolar TMM
8 (eq 1).
20 Various 1,1-2-alkylidene-dialkoxycyclopropanes can also be prepared by the same procedure from the corresponding 1,1-dialkoxy-2-alkylcyclopropanes, and have been shown to undergo regio- and stereoselective [3+2] cycloaddition reactions.
21 The use of a
nitrogen atmosphere and dry solvent is not essential, and the reactions can be carried out under air in the presence of a small amount of water (the use of a slight excess of the methylenecyclopropane removes any water without affecting the yield of the cycloaddition product). Solvent is also not essential; the reaction rate and course may be affected by the choice of solvent (or absence of solvent).
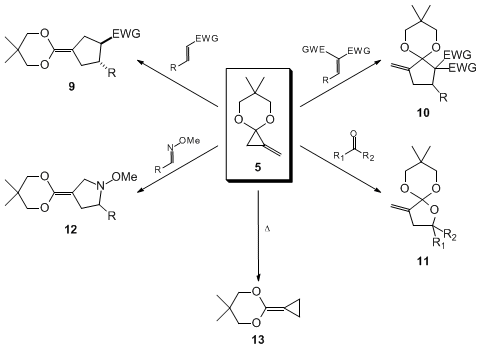
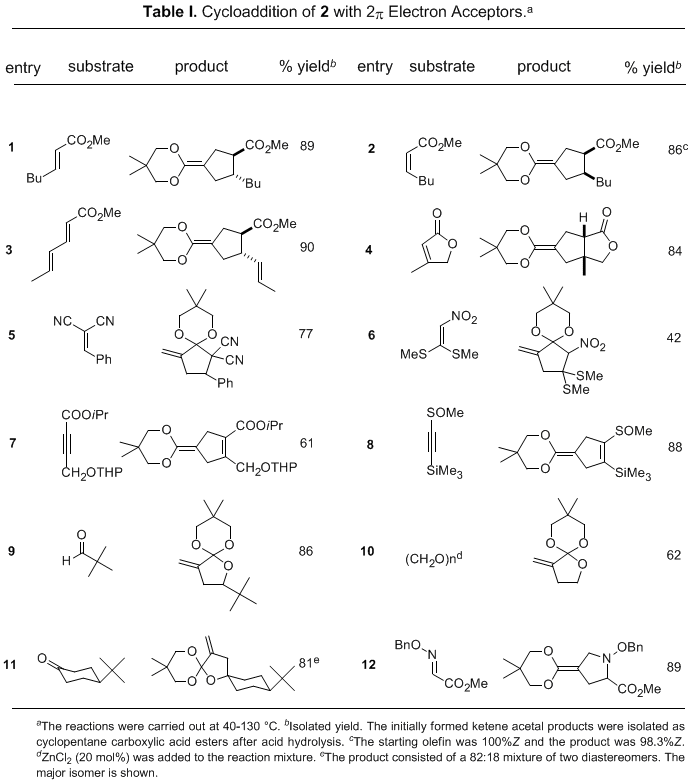
Olefins substituted with a single ester, nitrile, or ketone group function as good TMM acceptors to afford ketene acetals
9 (Table 1, entries 1-4). The cycloaddition reaction takes place stereoselectively with retention of the olefin geometry and "endo" orientation of the directing groups.
21 In contrast, the reaction with olefins whose reduction potential is larger than −1.8 eV (vs. SCE) gives mainly the acetal of an α-methylenecyclopentanone
10 via a single electron transfer process.
22 This cycloaddition can be used for the functionalization of buckminsterfullerene (C
60 and C
70)
23,24 whose reduction potential is −0.42 eV (see below). A radioactive version of
3and
5 has been prepared by using
14CH
3I in Step B and has been employed for pharmacokinetic studies of water soluble C
60.
25 Cycloaddition to electron-deficient acetylenes takes place smoothly to give cyclopentene carboxylic esters in one step (entries 7 and 8).
26 Cycloaddition to carbonyl compounds gives tetrahydrofuran derivatives (entries 9-11),
27 and to an O-alkyloxime affords a pyrrolidine (entry 12).
28 Prolonged heating gives a highly reactive ketene acetal
13, which serves as a useful synthon of cyclopropanecarboxylate enolate.
29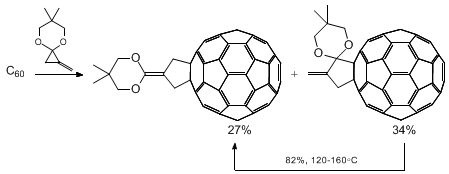
Appendix
Chemical Abstracts Nomenclature (Collective Index Number);
(Registry Number)
2,2-Bis(chloromethyl)-5,5-dimethyl-1,3-dioxane:
1,3-Dioxane, 2,2-bis(chloromethyl)-5,5-dimethyl- (9); (133961-12-3]
2-Cyclopenten-1-one (8, 9); (930-30-3]
1,3-Dichloroacetone:
2-Propanone, 1,3-dichloro- (8, 9); (534-07-6)
6,6-Dimethyl-1-methylene-4,8-dioxaspiro[2.5]octane:
4,8-Dioxaspiro[2.5]octane, 6,6-dimethyl-1-methylene (9); (122968-05-2)
Ferric nitrate nonahydrate:
Nitric acid, iron(3+) salt, nonahydrate (9); (7782-61-8)
Neopentyl glycol:
1,3-Propanediol, 2,2-dimethyl- (8, 9); (126-30-7)
Potassium tert-butoxide:
2-Propanol, 2-methyl-, potassium salt (9); (865-47-4)
Ammonia (8,9); (7664-41-7)
Sodium (8, 9); (7440-23-5)
Sodium amide:
Sodium amide [Na(NH2)] (9); (7782-92-5)
p-Toluenesulfonic acid:
Benzenesulfonic acid, 4-methyl- (9); (104-15-4)
1,6,6-Trimethyl-4,8-dioxaspiro[2.5]oct-1-ene:
4,8-Dioxaspiro[2.5]oct-1-ene, 1,6,6-trimethyl- (9); (122762-81-6)
Methyl iodide:
Methane, iodo- (8, 9); (74-88-4)
Copyright © 1921-, Organic Syntheses, Inc. All Rights Reserved