Org. Synth. 2005, 81, 235
DOI: 10.15227/orgsyn.081.0235
THE USE OF POLYSTYRYLSULFONYL CHLORIDE RESIN AS A SOLID SUPPORTED CONDENSATION REAGENT FOR THE FORMATION OF ESTERS: SYNTHESIS OF N-[(9-FLUORENYLMETHOXY)CARBONYL]-L-ASPARTIC ACID; α tert-BUTYL ESTER, β-(2-ETHYL[(1E)-(4-NITROPHENYL)AZO]PHENYL]AMINO]ETHYL ESTER
Submitted by Norbert Zander
1 and Ronald Frank
2.
Checked by Richard V. Coelha, Jr., Klaas Schildknegt, and Sarah E. Kelly.
1. Procedure
To a 50-mL polypropylene vial (Note 1) are added 0.839 g (2.67 mmol) of 2-[ethyl[4-[(1E)-(4-nitrophenyl)azo]phenyl]amino]ethanol (Disperse Red 1, Note 2), 0.985 g of (2.39 mmol) N-[(9H-fluoren-9-ylmethoxy)carbonyl]-L-aspartic acid,1-(1, 1-dimethylethyl) ester (Fmoc-L-Asp-OtBu, Note 3), 3.26 g (4.73 mmol) of polystyrylsulfonyl chloride resin (Note 4), and 30 mL anhydrous methylene chloride (Note 5). The vial is capped and the mixture is shaken for five min (Note 6). N-Methylimidazole (0.764 mL, 9.58 mmol) is then added to the deep red mixture (Note 7) and the resulting mixture is shaken for 2 hr (Note 8).
N-Methylimidazole is then removed from the reaction mixture with Amberlyst 15 ion exchange resin (Note 9) using the following procedure. To a 2-cm diameter column equipped with a glass frit (Note 1) is added 7 g of Amberlyst 15, which is rinsed with 25 mL of methylene chloride. The reaction mixture is added and the resin mixture is rinsed with 50 mL of methylene chloride. The filtrate is collected in a 250-mL flask (Note 10) and the solvent is removed on a rotary evaporator to afford 1.63-1.66 g (96-98%) of the desired ester 1 as a deep red foam. HPLC analysis showed a purity of 98% (Notes 11, 12, 13, 14).
2. Notes
1.
Polystyrene resin strongly sticks to glass, and consequently either
polypropylene reaction vessels or
silanized glassware should be used. Glassware was silanized by the following procedure. The glassware and a
small beaker containing neat
chlorotrimethylsilane were placed in a
desiccator. Vacuum was applied until the
chlorotrimethylsilane started to boil and then the desiccator was closed. The next day, the glassware was removed, rinsed with
methylene chloride, and dried at 80°C.
2.
Disperse Red 1, dye content ca. 95%, is available from Aldrich, Sigma-Aldrich Chemie GmbH. The checkers obtained
Disperse Red 1 from Sigma-Aldrich Corporation.
3.
The submitters obtained
Fmoc-L-Asp-OtBu from Calbiochem-Novabiochem AG. The checkers obtained
Fmoc-L-Asp-OtBu from EMD Biosciences, Inc.4.
Polystyrylsulfonyl chloride resin is available from Argonaut Technologies. The batch used by the submitters had a substitution of 1.44 mmol/g (the checker's batch had a substitution of 1.45 mmol/g). The use of
polystyrylsulfonyl chloride resin from Novabiochem in this type of reaction resulted in drastically longer reaction times and less pure products.
5.
Methylene chloride was the solvent most compatible with the reagents and resin.
Tetrahydrofuran or a
1 : 1 mixture of dimethylformamide (DMF) and methylene chloride could also be employed, but longer reaction times were necessary.
1,4-Dioxane,
toluene,
N-methylpyrrolidinone, and DMF alone were not suitable as solvents.
6.
Mixtures containing polystyrene resins should either be shaken or stirred with a
mechanical stirrer. Stirring with a
magnetic stir bar results in destruction of resin beads and the resulting debris can clog frits during filtrations.
7.
In place of
N-methylimidazole (Melm), only
dimethylaminopyridine (DMAP) could be substituted. The solid-supported amines piperidinomethyl- or morpholinomethyl polystyrene resins,
pyridine, and tertiary amines like
triethylamine and
N-methylmorpholine were not effective.
8.
HPLC analysis showed complete conversion after 90 min. The use of less base resulted in longer reaction times.
9.
Amberlyst 15 is available from Fluka, Sigma-Aldrich Chemie GmbH. The checkers obtained
Amberlyst 15 from Sigma-Aldrich Corporation. The use of only
6 g of Amberlyst 15 resulted in incomplete removal of MeIm.
10.
Further rinsing with
methylene chloride yielded trace quantities of additional product, which was less pure by HPLC analysis. The main impurity was
2 (see Discussion) produced by cleavage of the
t-butyl ester by the Amberlyst 15 resin.
11.
Analytical reversed-phase HPLC was performed using a 50 mm × 2 mm i.d. 3 µm C18(2) column (LUNA, Phenomenex, Germany); solvent A was 0.1% TFA in HPLC-grade water; solvent B was 0.1% TFA in HPLC-grade
acetonitrile; UV-detection was at 220 nm; flow rate was 0.8 mL/min; gradient elution: 0 min 95% A, 0-11 min 5% A, 11-12.5 min 5% A, 12.5-13 min 95% A, 13.5-17 min 95% A.
12.
The submitters performed the reaction using a
0.12 mmol excess of Fmoc-L-Asp-Ot-Bu, under which conditions aminomethylated polystyrene resin was required to remove the excess carboxylic acid
(Note 13). The checkers modified the reaction to use
0.28 mmol excess Disperse Red 1. The initial Amberlyst-15 filtration removes this material.
13.
To remove carboxylic acid, the crude product is redissolved in
20 mL of methylene chloride and is shaken for 30 min with 1 g of aminomethylated polystyrene resin with a substitution of 1.02 mmol/g, available from Novabiochem
(Note 3). After filtration and washing of the resin with
50 mL of methylene chloride, the filtrates were collected together in a
250-mL flask and the solvent was removed on a
rotary evaporator.
14.
Spectral properties of the product are as follows: IR (KBr): cm
−1 2975, 1733, 1600, 1549, 1514, 1433, 1391, 1336, 1253, 1133, 853, 741;
1H NMR (400 MHz, DMSO-d
6): δ 1.11 (t, 3H, J = 6.8 Hz), 1.30 (s, 9H), 2.61 (dd, 1H, J = 16.4, 8.1 Hz), 2.71 (dd, 1H, J = 16.2, 6.6 Hz), 3.50 (q, 2H, J = 6.9 Hz), 3.68 (m, 2H), 4.18 (t, 1H, J = 6.6 Hz), 4.32 – 4.23 (m, 5H), 6.88 (d, 2H, J = 9.5 Hz), 7.28 (t, 2H, J = 7.5 Hz), 7.37 (t, 2H, J = 7.3 Hz), 7.66 (d, 2H, J = 7.5 Hz), 7.74 (d, 1H, J = 8.3 Hz), 7.81 (d, 2H, J = 9.1 Hz), 7.85 (d, 2H, J = 7.5 Hz), 7.88 (d, 2H, J = 9.1 Hz), 8.32 (d, 2H, J = 9.1 Hz);
13C NMR (100 MHz, DMSO-d
6): δ 12.6, 28.1, 36.1, 36.7, 45.7, 47.2, 48.8, 51.6, 62.6, 66.3, 81.8, 112.4, 120.2, 120.8, 123.1, 123.8, 125.6, 125.8, 126.8, 127.7, 128.3, 136.3, 141.4, 143.4, 144.4, 147.5, 152.3, 156.5, 156.7, 170.5, 170.7; HRMS (ESI): Calcd for C
39H
41N
5O
8+Na
+: 730.2853, found 730.2854.
Handling and Disposal of Hazardous Chemicals
The procedures in this article are intended for use only by persons with prior training in experimental organic chemistry. All hazardous materials should be handled using the standard procedures for work with chemicals described in references such as "Prudent Practices in the Laboratory" (The National Academies Press, Washington, D.C., 2011 www.nap.edu). All chemical waste should be disposed of in accordance with local regulations. For general guidelines for the management of chemical waste, see Chapter 8 of Prudent Practices.
These procedures must be conducted at one's own risk. Organic Syntheses, Inc., its Editors, and its Board of Directors do not warrant or guarantee the safety of individuals using these procedures and hereby disclaim any liability for any injuries or damages claimed to have resulted from or related in any way to the procedures herein.
3. Discussion
Although solid-supported reagents and scavengers have been used in organic synthesis for decades, it was the development of combinatorial and parallel high throughput synthesis techniques that brought this class of reagents to wider attention. While there are numerous applications of solid-supported reagents and scavengers only a few examples for the formation of esters have been described.
3 Carboxylates, generated with solid-supported organic bases, were alkylated with alkyl halides.
4 Solid-supported organic bases were also used as scavenger resins in the esterification of
benzyl alcohol with benzoyl chlorides, giving clean benzyl esters in high yields.
5 This approach requires the acid chloride to be available. A recent report describes the alkylation of carboxylic acids with carbenium ions, generated from polymer-supported triazines.
6 This approach, however, requires the preformation of the polymer-supported triazine for each alkyl group to be transferred and a relatively high excess of the alkylating resin. No examples for the formation of aryl esters are given.
We report here a convenient and general procedure for the formation of esters directly from the easily available carboxylic acids and alcohols or phenols.
7 The reaction uses
polystyrylsulfonyl chloride resin as an efficient dehydration reagent and
N-methylimidazole as basic catalyst. The method employs only commercially available supported reagents and scavengers and allows compounds to be obtained in excellent yields and high purity by simple filtration. It should therefore be especially suitable for an automated parallel synthesis. The scope of our procedure was examined by treating six carboxylic acids with Fmoc-glycinol and seven alcohols with Fmoc-glycine. The results are summarized in Table 1. The purity of all the different esters derived from Fmoc-glycinol (entry 1-6) was very good (>93%), while the reaction time necessary for a complete conversion varied considerably. All products of the esterification of Fmoc-glycine (entry 7-13) with different alcohols and donor or acceptor substituted phenols were essentially 100% pure, except for
tert-butyl alcohol, which reacted only after the addition of 0.25 equivalents of DMAP. The conversion was slow, but the resulting ester was of good purity.
Scheme 1:
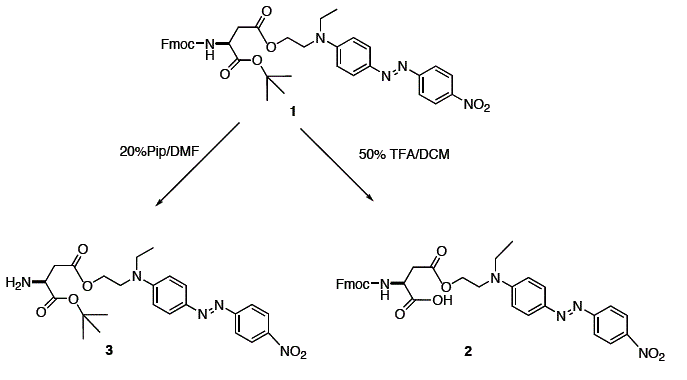
The example described in the experiment illustrates the compatibility of this reaction and the work up procedure with the base-sensitive Fmoc- as well as the acid-sensitive
tert-Bu-ester protecting group. No by-product
3 due to Fmoc cleavage by
N-methylimidazole was found. The small amount of by-product
2 resulting from the cleavage of the tert-butyl ester by the acidic ion exchange resin Amberlyst 15 during the removal of
N-methylimidazole is easily separated during this process
(Note 13). The product
1 has potential in the synthesis of color-labelled peptides
8 after cleavage of either of the protecting groups as illustrated in Scheme 1. The amine
3 should be useful for peptide synthesis in solution while the carboxylic acid
2 could be applied for solid phase synthesis. The Fmoc group was easily removed in 10 min with
20% piperidine in DMF. LC-MS analysis of
3 showed no by-products resulting from the cleavage of the two ester groups. The
tert-butyl ester was cleaved in one hour with 50% TFA in
methylene chloride without side reactions.
Appendix
Chemical Abstracts Nomenclature (Collective Index Number);
(Registry Number)
Disperse Red 1; Ethanol, 2-[ethyl[4-[4-nitrophenyl)azo]phenyl]amino]-; (2872-52-8).
N-[(9H-fluoren-9-ylmethoxy)carbonyl]-L-aspartic acid, 1-(1,1-dimethylethyl) ester;
L-Aspartic acid, N-[(9H-fluoren-9-ylmethoxy)carbonyl]-, 1-(1,1-dimethylethyl ester; (129460-09-9).
N-Methylimidazole:
1H-lmidazole, 1-methyl-; (616-47-7).
Copyright © 1921-, Organic Syntheses, Inc. All Rights Reserved