Org. Synth. 2005, 81, 262
DOI: 10.15227/orgsyn.081.0262
BORIC ACID CATALYZED AMIDE FORMATION FROM CARBOXYLIC ACIDS AND AMINES: N-BENZYL-4-PHENYLBUTYRAMIDE
[(Benzenebutanamide, N-(phenylmethyl)-)]
Submitted by Pingwah Tang
1
Checked by Helga Krause and Alois Fürstner.
1. Procedure
N-Benzyl-4-phenylbutyramide. A flame-dried, 200-mL, three-necked, round-bottomed flask is equipped with two glass stoppers, a vacuum-jacketed Dean-Stark trap topped with a reflux condenser fitted with a nitrogen inlet, and a Teflon-coated magnetic stirring bar. The reaction vessel is charged with 4-phenylbutyric acid (4.92 g, 30 mmol) (Note 1), boric acid (0.020 g, 0.30 mmol) (Note 2), and 88 mL of toluene (Note 3). To the stirred colorless reaction mixture is added benzylamine (3.32 g, 31 mmol) (Note 4) in one portion. The reaction mixture is heated at reflux for 16 hr and ca. 0.6 mL of water is collected in the Dean-Stark trap (Notes 5, 6, 7, 8). The mixture is allowed to cool to ambient temperature and then is poured with stirring into 500 mL of hexanes leading to the immediate precipitation of a white solid. Stirring is continued for an additional 30 min and then the precipitate is filtered off with suction through a sintered glass filter funnel. The collected solid is successively washed with two 60-mL portions of hexanes and two 60-mL portions of distilled water (Note 9) and then is dried in vacuo at room temperature for 12 hr to afford 6.90 g (91%) of N-benzyl-4-phenylbutyramide as a white solid (Notes 10, 11).
2. Notes
1.
4-Phenylbutyric acid (99%) was purchased from Aldrich Chemical Company and used as received.
2.
Boric acid (99.999%) was purchased from Aldrich Chemical Company and used as received.
3.
Toluene (99.8% anhydrous, water <0.001%, evaporation residue <0.005%) was purchased from Aldrich Chemical Company and used as received. The capacity of the receiver in the
Dean-Stark trap was 28 mL. The initial amount of
toluene placed in the flask was 88 mL. Upon heating to reflux,
28 mL of toluene was distilled from the flask and collected in the receiver. The remaining volume of
toluene in the reaction flask was ca. 60 mL, corresponding to an approximately 0.50M concentration of the reactants.
4.
Benzylamine (redistilled 99.5+ %) was purchased from Aldrich Chemical Company and used without further purification.
5.
The theoretical amount of water is 0.54 mL.
6.
TLC analysis indicated that the reaction was complete. TLC was performed on pre-coated plates with
silica gel 60 F254 purchased from EM Sciences (Merck) using
hexanes/ethyl acetate (1/1) as the eluent.
7.
HPLC analysis indicated that the reaction was complete. HPLC was conducted utilizing Perkin-Elmer's Series 200 autosampler and pump with either a Perkin-Elmer Series 200 or Applied Biosystems 785A UV/Vis detector (both monitoring at (λ = 220 nm). Phenomenex's Kromasil 5 micron C18 column (50 mm × 4.6 mm) fitted with Phenomenex's C18 (ODS, Octadecyl) security guard cartridges (4 mm × 3 mm) were utilized with a 3 mL/min flow-rate. The solvent system consisted of mixtures of
acetonitrile and water, containing 0.1% (v/v)
trifluoroacetic acid, with the gradient starting at 5%
acetonitrile and increasing to 95% over 10.5 min.
8.
GC analysis indicated that the reaction was complete. Gas chromatographic analyses were performed on a Agilent 6890N GC system equipped with a 30-m 5%
polyphenyl methyl siloxane capillary column (0.32 mm i.d.). Helium was used as the carrier gas, and the flow rate was kept constant at 7.7 mL/min. The retention time was measured under the following conditions: injector temperature: 250°C; initial temperature of the column: 150°C; increment rate 8°C/min to 200°C. After being kept at 200°C for 8.0 min, the temperature was raised to 290°C at an increment rate of 8°C/min.
9.
The water wash removes residual
boric acid. Elemental analysis of the final product indicated that
boron content was less than 0.005%.
10.
The filtrate (wash liquor) contained some white solid which was collected, triturated with
ca. 5 mL of heptane, and dried to provide additional 0.35-0.50 g of pure product as a white solid (0.4-0.7%). Analytical data (TLC, GC, HPLC and elemental analysis) indicated that this crop has a purity of ≥95%.
11.
The amide was obtained in
88-91% yield in different runs. The product has the following physico-chemical properties: GC: R
t: 10.31 min
(Note 8); HPLC: R
t: 5.63 min
(Note 7); TLC R
f: 0.40
(Note 6);
mp 78-80°C; IR (Kbr:) cm
−1 3293, 3085, 3029, 2944, 1632, 1550, 1453, 1236, 1155, 1080, 1027, 724, 695;
1H NMR (300 MHz, DMSO-d
6): δ 1.76-1.87 (m, 2 H), 2.15 (t, 2 H, J = 7.2), 2.55 (t, 2 H, J = 7.5), 4.26 (d, 2 H, J = 6.0), 7.16-7.33 (m, 10 H), 8.30 (br, 1 H);
13C NMR (75.6 MHz, DMSO-d
6): δ 27.1, 34.6, 34.7, 41.9, 125.7, 126.6, 127.1, 128.20, 128.23, 128.27, 139.7, 141.7, 171.7. Anal. calcd for C
17H
19NO: C, 80.60; H, 7.56; N, 5.53. Found: C, 80.29; H, 7.53; N, 5.47; B < 0.005.
Handling and Disposal of Hazardous Chemicals
The procedures in this article are intended for use only by persons with prior training in experimental organic chemistry. All hazardous materials should be handled using the standard procedures for work with chemicals described in references such as "Prudent Practices in the Laboratory" (The National Academies Press, Washington, D.C., 2011 www.nap.edu). All chemical waste should be disposed of in accordance with local regulations. For general guidelines for the management of chemical waste, see Chapter 8 of Prudent Practices.
These procedures must be conducted at one's own risk. Organic Syntheses, Inc., its Editors, and its Board of Directors do not warrant or guarantee the safety of individuals using these procedures and hereby disclaim any liability for any injuries or damages claimed to have resulted from or related in any way to the procedures herein.
3. Discussion
Many procedures for the formation of carboxylic acid amides are known in the literature. The most widely practiced method employs carboxylic acid chlorides as the electrophiles which react with the amine in the presence of an acid scavenger. Despite its wide scope, this protocol suffers from several drawbacks. Most notable are the limited stability of many acid chlorides and the need for hazardous reagents for their preparation (
thionyl chloride,
oxalyl chloride,
phosgene etc.) which release corrosive and volatile by-products. Moreover, almost any other functional group in either reaction partner needs to be protected to ensure chemoselective amide formation.
2 The procedure outlined above presents a convenient and catalytic alternative to this standard protocol.
Although there are several reports in the literature on boron-mediated amide formations, the boron reagents had to be used in stoichiometric amounts.
3,4,5,6,7,8,9 Recently, Yamamoto et al. presented the first truly catalytic method allowing for a direct amide formation from free carboxylic acids and amines as the reaction partners.
10,11,12 Best results were obtained by using phenylboronic acids bearing electron withdrawing substituents in the
meta- and/or
para-positions such as
3,4,5-trifluorophenylboronic acid or
3,5-bis(trifluoromethyl)boronic acid as the catalysts.
During the course of our discovery program directed to small molecules for drug delivery, it was discovered that cheap, readily available, non-toxic, and environmentally benign boric acid, B(OH)3, also constitutes a highly effective catalyst for direct amide formation. Benzylamines and cyclic aliphatic amines such as piperidines react smoothly. In most cases, the use of 5 mol% of B(OH)3 is sufficient for obtaining excellent yields. Likewise, aniline derivatives afford the corresponding amides without incident even if they are hardly nucleophilic due to the presence of electron withdrawing substituents on the arene ring; in such cases, however, the amount of B(OH)3 has to be increased to ca. 25% to ensure complete conversion. The scope of the method is illustrated by the examples compiled in Tables 1 and 2. Particularly noteworthy are the operational simplicity of this new method which might therefore qualify for large-scale preparations, as well as the excellent chemoselectivity profile that can make protection/deprotection sequences obsolete. This is illustrated by the examples shown below.
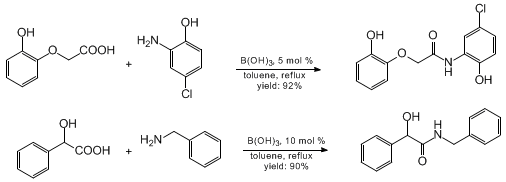
It is proposed that the
boric acid reacts with the carboxylic acid to form a mixed anhydride as the actual acylating agent.
9,13 Upon reaction with an amine, this intermediate forms the desired carboxamide and regenerates the catalytically active
boric acid.
Several important generalizations emerge from the study of the boric acid catalyzed amidation of carboxylic acids and amines.
- In most cases, 5 mol% of the catalyst is sufficient to catalyze the amidation. In all cases, the reactions proceed cleanly in high yields to the expected carboxamides.
- No or little side reactions either with the unprotected hydroxy group present in the phenylamines, or with the unprotected hydroxyl group present in the carboxylic acids are observed. The catalytic amidation of unprotected α-hydroxycarboxylic acids also proceeds well under similar conditions.
- The present method is successfully applicable to aromatic amines, which are less nucleophilic and less reactive. It works well with phenylamines, even bearing deactivating groups such as carboxylic acid ester moieties; in these cases we use 25 mol% of boric acid to drive the reaction to completion.
- The catalyst employed in the reactions, boric acid, is inexpensive and commercially available. Boric acid is a "green" catalyst. By virtue of the simplicity of the process, the operation is easy to conduct. Therefore, it is amenable for large-scale preparations.
- The catalytic amidation is an atom-economical process because it maximizes the incorporation of all materials used in the process into the final product. Therefore, it allows organic molecule architects for the quick building of molecular complexity.
Appendix
Chemical Abstracts Nomenclature (Collective Index Number);
(Registry Number)
N-Benzyl-4-phenylbutyramide:
Benzenebutanamide, N-(phenylmethyl)-; (179923-27-4)
4-Phenylbutyric acid:
Benzenebutanoic acid; (1821-12-1)
Boric acid:
Boric acid (H3BO3); (10043-35-3)
Benzylamine:
Benzenemethanamine; (100-46-9)
Copyright © 1921-, Organic Syntheses, Inc. All Rights Reserved