Org. Synth. 2005, 82, 10
DOI: 10.15227/orgsyn.082.0010
PREPARATION OF OPTICALLY ACTIVE (R,R)-HYDROBENZOIN FROM BENZOIN OR BENZIL
[1,2-Ethanediol, 1,2-diphenyl-, (1R,2R)-]
Submitted by Takao Ikariya
1, Shohei Hashiguchi
2, Kunihiko Murata
3, and Ryoji Noyori
4.
Checked by Peter Wipf and David Amantini.
1. Procedure
A. A 100 g Scale Synthesis from rac-Benzoin: A 1 L four-necked, round-bottomed flask equipped with a mechanical stirrer, a reflux condenser bearing an inert gas inlet tube, a thermometer and a dropping funnel is charged with 290 mL (2.08 mol) of triethylamine (Note 1). The triethylamine is cooled to 4°C in an ice bath and formic acid (97.0 mL, 2.57 mol) is added slowly (Note 2). To the mixture of formic acid and triethylamine at ambient temperature is added rac-benzoin (Note 3) (170 g, 0.801 mol), RuCl[1S,2S)-N-p-toluenesulfonyl-1,2-diphenylethanediamine]-(η6-p-cymene) (Notes 4 and 5) (0.204 g, 0.321 mmol), and dry DMF (Notes 1 and 6) (80 mL). After the reaction mixture is stirred at 40°C for 48 h, 300 mL of water is added at 0°C with stirring (Note 7). The pale pink precipitate is filtered through a Büchner funnel, washed with water (2 × 500 mL), and dried in vacuo to give a white solid in 97% yield (Note 8). The crude product is dissolved in hot methanol (700 mL) at 60°C. A small amount of insoluble material is removed through filtration and the filtrate is cooled initially to room temperature and then to 0 to 5°C to provide white crystals. The crystalline product is isolated by filtration, washed with cooled (ice bath) 2-propanol (400 mL), and dried to provide 129.7 g of optically pure (R,R)-hydrobenzoin as white crystals (dl > 99%, 99.9% ee, (Note 9)). Concentration of the mother liquors and another recrystallization from methanol (100 mL) provides a second crop of the product, 19.1 g (dl > 99%, 99.9% ee, (Note 9)). The overall yield is 148.8 g (87%).
B. A 10 g Scale Synthesis from Benzil: A 100 mL four-necked, round-bottomed flask equipped with a mechanical stirrer, a reflux condenser bearing an inert gas inlet tube, a thermometer, and a dropping funnel is charged with a mixture of formic acid (8.70 mL, 230 mmol) and triethylamine (19.0 mL, 136 mmol) in a similar manner to Procedure A. To this formic acid-triethylamine mixture at ambient temperature is added benzil (Note 3) (11.0 g, 52.3 mmol), and RuCl[(1S,2S)-N-p-toluenesulfonyl-1,2-diphenylethanediamine](η6-p-cymene) (33.3 mg, 0.0524 mmol) (Note 6). After the reaction mixture is stirred at 40°C for 24 h, 50 mL of water is added at 0°C with stirring. The pale pink precipitate is filtered through a Büchner funnel, washed with water (50 mL), and dried in vacuo to give a white solid in 95% yield. The crude product is dissolved in hot methanol (50 mL) at 60°C. The filtrate is cooled to room temperature and then to −40°C to give white crystals. The crystalline product is isolated by filtration, washed with cooled (ice-bath) 2-propanol (10 mL), and dried to provide 9.3 g of optically pure (R,R)-hydrobenzoin as white crystals (82%, 100% ee).
2. Notes
1.
Triethylamine and formic acid were purchased from Kanto Chemical Company and used without further purification. The Checkers used chemicals from Fisher and Fluka. An azeotropic mixture of
triethylamine and
formic acid is commercially available but could not be used for this reaction (see discussion section).
Dry DMF was purchased from J. T. Baker.
2.
The reaction of
triethylamine with
formic acid is exothermic and may proceed violently unless performed by controlled addition.
3.
Benzoin and benzil were purchased from Kanto Chemical Company and used without further purification. The Checkers used chemicals from TCI and Acros, respectively. Substituted benzoins were prepared by
benzoin condensation of the corresponding ring-substituted benzaldehydes.
54.
The Checkers used the following procedure for the preparation of
(1S,2S)-N-p-toluenesulfonyl-1,2-diphenylethanediamine (TsDPEN): a dry
CH2Cl2 solution
(10 mL) of p-toluenesulfonyl chloride (0.893 g, 4.69 mmol) was added dropwise over 5 h (syringe pump addition) to a mixture of
(S,S)-DPEN (0.995 g, 4.69 mmol) and
triethylamine (0.69 mL, 4.5 mmol) in dry
CH2Cl2 (30 mL) at 0°C. After the reaction mixture was stirred at 0°C for 6 h, the solution was washed with water (10 mL × 2) and saturated
NaCl solution (10 mL) and then dried with
Na2SO4. The solvent was removed under reduced pressure to give
1.659 g of crude white solid product. Recrystallization from
ethyl acetate (8 mL) gave
1.146 g (
67% yield) of the desired product as white crystals.
5.
Commercially available chiral
Ru complexes from Kanto Chemical Company were used by the Submitters; however, the complexes were prepared by the Checkers following a modified literature procedure.
6 (R)-RuCl[(1S,2S)-p-TsNCH(C6H5)CH(C6H5)NH2](η6-p-cymene): A mixture of
[RuCl2(η6-p-cymene)]27 (0.651 g, 1.06 mmol), (S,S)-TsDPEN
8 (0.780 g, 2.13 mmol), and
triethylamine (0.60 mL, 4.3 mmol) in
2-propanol (21 mL) was stirred at 80°C for 1 h. The orange solution was concentrated and the resulting solid was collected by filtration, washed with a small amount of water and dried under reduced pressure to give the chiral Ru complex. After recrystallization from
methanol (20 mL),
0.552 g of pure Ru (II) catalyst were collected as bright orange crystals. After two additional recrystallizations of the concentrated mother liquor from
methanol (8.0 and 5.0 mL, respectively), an additional
0.327 g of pure catalyst were collected. The overall yield was
0.879 g (
65%).
mp >100°C (dec.); IR (KBr) [cm
−1]: 3468, 3277, 3220 (H–N), 3062, 3029 (H–C
aromat.), 2961, 2872 (H–C
aliphat.); MS (EI):
m/z (%) = 603 (63), 601 (100), 600 (66), 599 (58); 1H NMR
pdf (300 MHz, CDCl
3, 25°C, TMS): δ 1.37 (m, 6H, CH(CH
3)
2), 2.21 (s, 3H, CH
3 in p-cymene), 2.35 (s, 3H, CH
3 in
p-Ts), 3.12 (m, 1H, CH(CH
3)
2), 3.52 (m, 2H, N
HH and
HCNH
2), 3.73 (d, 1H,
J = 10.5 Hz,
HCN-
p-Ts), 5.72 (m, 4H, CH
aromat. in p-cymene), 6.04 (m, 1H, NH
H), 6.34–7.05 (m, 14H,
p-CH
3C
6H
4-SO
2NCH(C
6H
5)CH(C
6H
5)NH
2).
6.
DMF was used to maintain the homogeneity of the reaction mixture, but it is not crucial for the catalysis to be efficient and practical. On small scale the addition of DMF is not needed. In fact, the reaction of
benzil with a substrate to catalyst ratio (S/C) of 1,000 (4.7 M) in a mixture of
HCOOH and N(C
2H
5)
3 containing the
(S,S)-Ru catalyst (
benzil:HCOOH:N(C
2H
5)
3 = 1:4.4:2.6) proceeded heterogeneously at the early stages of the reaction because of the low solubility of
benzil. After about ten minutes, the reaction mixture changed to a completely homogenous solution, giving almost the same results as with DMF as solvent.
7.
On large scale, the reaction flask should be connected to an Ar gas inlet to allow
CO2 to escape.
8.
The diastereoselectivity of the product,
dl:
meso = 95.0:5.0, was determined by integration in the 1H NMR
pdf (300 MHz, CDCl
3); (
R,
R)-hydrobenzoin: δ 3.07 (s, 2H, OH), 4.86 (s, 2H, CH–OH), 7.25–7.40 (m, 10H, aromatic ring protons),
meso-hydrobenzoin: δ 2.33 (s, 2H, OH), 4.98 (s, 2H, CHOH), 7.30–7.45 (m, 10H, aromatic ring protons).
9.
(R,R)-Hydrobenzoin:
[α]D25 +91.6 (c 1.05, ethanol), (lit.
9a [α]D23, +95 (c 0.87 ethanol), 99% ee (
R,
R)). HPLC separation conditions, (column: CHIRALCEL OJ (4.6 mm i.d. × 250 mm), eluent:
hexane/
2-propanol = 90/10, flow rate: 1.0 mL/min, temp: 25°C, detection UV 254 nm); retention time, (
S,S)-hydrobenzoin, 14.2 min,
(R,R)-hydrobenzoin, 16.5 min.
Handling and Disposal of Hazardous Chemicals
The procedures in this article are intended for use only by persons with prior training in experimental organic chemistry. All hazardous materials should be handled using the standard procedures for work with chemicals described in references such as "Prudent Practices in the Laboratory" (The National Academies Press, Washington, D.C., 2011 www.nap.edu). All chemical waste should be disposed of in accordance with local regulations. For general guidelines for the management of chemical waste, see Chapter 8 of Prudent Practices.
These procedures must be conducted at one's own risk. Organic Syntheses, Inc., its Editors, and its Board of Directors do not warrant or guarantee the safety of individuals using these procedures and hereby disclaim any liability for any injuries or damages claimed to have resulted from or related in any way to the procedures herein.
3. Discussion
Optically active hydrobenzoins are useful building blocks for the stereoselective synthesis of various biologically active compounds, as well as chiral ligands and auxiliaries. The preparation of these chiral hydrobenzoins by Sharpless asymmetric dihydroxylation of
trans-stilbene is one of the most convenient and well established methods.
9 Asymmetric reduction of readily available benzils or benzoins would be a promising and widely applicable approach; however, no practical reduction systems have been reported except for
oxazaborolidine-catalyzed reductions of benzils with
borane/methylsulfide.
10 Direct asymmetric hydrogenation of benzils or benzoins catalyzed by well-established Ru-BINAP complexes could potentially lead to optically active 1,2-diols. However,
meso-isomers are obtained as the major products, because the substrate control of the hydroxy ketone intermediate favors
meso-diol formation.
11 The procedure described herein provides a highly efficient method accessing the desired chiral hydrobenzoins in high enantiomeric excess using commercially available chiral Ru(II)catalysts, RuCl(TsDPEN)(η
6-arene),
6,12 and easily handled reagents such as benzils or benzoins as substrates and a
formic acid and
triethylamine mixture as the
hydrogen source.
13A mixture of formic acid and triethylamine is the best hydrogen donor for this reduction. In the absence of triethylamine, no conversion of benzoins or benzils was observed. The addition of triethylamine to the reaction mixture causes a significant increase in the conversion of the substrates. In the reaction of benzoin, a formic acid:triethylamine molar ratio of 3.2:2.6 to 3.2:4.4 gives the best catalyst performance in terms of both reactivity and stereoselectivity. The reduction of benzil requires a molar ratio of 4.4:2.6 to 4.4:4.4. The reaction with an azeotropic mixture of formic acid and triethylamine (5:2) gave no conversion under otherwise identical conditions as described in the Procedure.
The success of this asymmetric reduction of
benzil or
benzoin leading to the optically active
hydrobenzoin with the
formic acid and
triethylamine mixture relies strongly on the nature of
benzoin with a configurationally labile stereogenic center and the enantiomer discrimination ability of the chiral Ru complexes. Due to a sufficiently rapid stereomutation of benzoins under the basic reaction conditions, the dynamic kinetic resolution of benzoins allows the diastereo- and enantioselective synthesis of optically active hydrobenzoins.
13 Reduction of
(R)-benzoin with the
(S,S)-Ru catalyst in DMF under the same conditions gave
(R,R)-hydrobenzoin quantitatively and in 100% ee, indicating that the
(S,S)-Ru catalyst favors the reaction of
(R)-benzoin.
13a The rate of the reduction of
(R)-benzoin with the
(S,S)-Ru catalyst proceeds 55 times faster than the
S-isomer. The slow-reacting
S-isomer undergoes a rapid racemization.
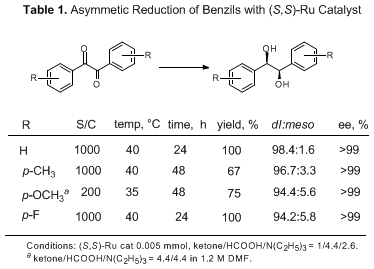
Various benzil derivatives bearing substituents on aromatic rings can be reduced stereoselectively to the chiral hydrobenzoins in high ee's and in good yields (Table 1). The benzils with electron-donating substituents such as methyl or methoxy groups are reduced with excellent enantioselectivity but more slowly, while the reduction of
p-fluorobenzil proceeded rapidly, as expected, giving a product with a high ee.
13The described, chiral Ru catalyst-promoted asymmetric transfer hydrogenation with a
formic acid and
triethylamine mixture is also applicable to the enantioselective reduction of
acetophenone,
12 ring-substituted
acetophenone derivatives,
12 α-substituted acetophenones,
14,15 acetylpyridine derivatives,
16 and functionalized ketones
17,18 leading to the corresponding optically active alcohols in excellent ee. These asymmetric reductions with the chiral Ru catalyst are characterized by a rapid, carbonyl group-selective transformation because of the coordinatively saturated nature of the diamine-based Ru hydride complexes.
6,17 The neighboring groups at the α-position of the carbonyl group do not interact with the metal center, leading to excellent reactivity and enantioselectivity. Some representative examples are listed in Figure 1.
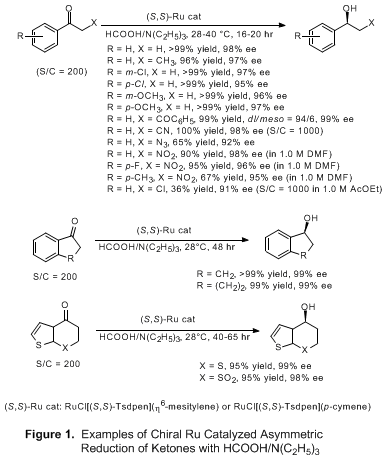
Appendix
Chemical Abstracts Nomenclature (Collective Index Number);
(Registry Number)
Formic acid; (64-18-6)
Triethylamine:
Ethanamine, N,N-diethyl-; (121-44-8)
rac-Benzoin:
Ethanone, 2-hydroxy-1,2-diphenyl-; (19-53-9)
RuCl[(1S,2S)-p-TsNCH(C6H5)CH(C6H5)NH2](η6-p-cymene):
Ruthenium, [N-[(1S,2S)-2-(amino-κN)-1,2-diphenylethyl]-4-methyl-benzenesulfonamidato-κN]chloro[(1,2,3,4,5,6-η)-1-methyl-4-(1-methylethyl)benzene]-; (192139-90-5)
(R,R)-Hydrobenzoin:
1,2-Ethanediol, 1,2-diphenyl-, (1R,2R)-; (52340-78-0)
2-Propanol; (67-63-0)
Benzil:
Ethanedione, diphenyl-; (134-81-6)
Copyright © 1921-, Organic Syntheses, Inc. All Rights Reserved