Org. Synth. 2007, 84, 58
DOI: 10.15227/orgsyn.084.0058
SYNTHESIS OF AN ANTI-α-AMINO EPOXIDE BY ONE-CARBON HOMOLOGATION OF AN α-AMINO ESTER:
[(2S,3S)-1,2-EPOXY-3-(BOC-AMINO)-4-PHENYLBUTANE]
Submitted by Dengjin Wang
1a and William A. Nugent.
1b
Checked by Jason J. Kowal and David J. Mathre.
1. Procedure
A. Dimethylsulfoxonium (S)-2-oxo-3-(Boc-amino)-4-phenylbutylide (2). A 500-mL three-necked, round-bottomed flask is equipped with a large stir bar, a nitrogen inlet, a reflux condenser capped with a nitrogen outlet, and a rubber septum through which is inserted a temperature probe. The flask is flushed with nitrogen for 5 min and is then charged with trimethylsulfoxonium iodide (33.0 g, 150 mmol) (Note 1), which is added through a neck that is temporarily opened, and 150 mL of anhydrous tetrahydrofuran (Note 2), which is added by syringe through a septum. A 1.0 M solution of potassium tert-butoxide in tetrahydrofuran (150 mL, 150 mmol) (Note 3) is added via syringe through a septum, after which the mixture is heated at reflux for 2 h (Note 4). The resulting solution of dimethylsulfoxonium methylide (containing precipitated potassium iodide) is cooled to 0 °C. The reflux condenser is replaced with a 125-mL pressure-equalizing addition funnel containing a solution of Boc-L-phenylalanine 4-nitrophenyl ester (1) (19.3 g, 50.0 mmol) (Note 5) in 100 mL of anhydrous tetrahydrofuran and capped with a nitrogen outlet. The solution is added dropwise over 20 min so that the internal temperature does not exceed 5 °C. The mixture is stirred for 1 h at 0 °C. The flask is briefly opened by temporary removal of the addition funnel to allow addition of 50 mL of water and stirring is continued for 15 min at 0 °C. After warming to room temperature, the mixture is filtered through a short pad (3 mm) of Celite on a 100-mL fritted-glass Büchner funnel and the solvent is removed by rotary evaporation (25 °C, 20 mmHg). The concentrated mixture is rinsed into a 1-L separatory funnel with 200 mL of water and extracted with one 200-mL portion and two 100-mL portions of ethyl acetate (Note 6). The combined extracts are washed with one 50-mL portion of water and two 50-mL portions of brine. (In some cases solid product separates during the first extraction, but it redissolves when the organic phases are combined.) The solution is dried over sodium sulfate, filtered and concentrated by rotary evaporation (25 °C, 20 mmHg) to afford 2 as a light yellow solid (15.3–15.4 g, 90–91%) (Note 7), which is carried on to the next step without further purification.
B. (S)-1-Chloro-3-(Boc-amino)-4-phenyl-2-butanone (3). A 500-mL three-necked, round-bottomed flask is equipped with a stir bar, a nitrogen inlet, a reflux condenser capped with a nitrogen outlet, and a rubber septum through which is inserted a temperature probe. The flask is flushed with nitrogen for 5 min and is then charged with dimethylsulfoxonium (S)-2-oxo-3-(Boc-amino)-4-phenylbutylide (2) (13.6 g, 40 mmol), which is added through temporary removal of the reflux condenser. Anhydrous tetrahydrofuran (250 mL) is added by syringe through a septum. A 4.0 M solution of hydrogen chloride in 1,4-dioxane, (11.6 mL, 46.0 mmol) (Note 8) is added via syringe through the septum over 5 min at room temperature during which time a solid sulfoxonium salt separates. The mixture is then heated with stirring at reflux (70 °C) for 4 h. The reaction mixture stirs as a very thick slurry for the first 30 min of this period. After cooling to room temperature, the resulting homogeneous solution is transferred to a 1-L separatory funnel, to which is added 100 mL of ethyl acetate and 200 mL of hexanes (Note 9). The mixture is washed with two 200-mL portions of water, one 100-mL portion of saturated sodium bicarbonate solution, four 200-mL portions of water and one 100-mL portion of brine. (Note 10) The solution is dried over sodium sulfate and filtered through a 100-mL fritted-glass Büchner funnel. The filtrate is concentrated by rotary evaporation (25 °C, 20 mmHg) to obtain 13–14 g of a light yellow solid. This material is dissolved in 40 mL of hot hexanes (70 °C). Upon cooling, a solid separates which is collected by filtration on a 100-mL fritted-glass Büchner funnel and dried overnight at 25 °C and 0.5 mmHg to afford 3 (9.61–9.71 g, 81% yield) (Note 11) as an off-white solid.
C. (2S,3S)-1,2-Epoxy-3-(Boc-amino)-4-phenylbutane (4). A 500-mL three-necked, round-bottomed flask is equipped with a large stir bar (Note 12), a nitrogen inlet, a 125-mL pressure-equalizing addition funnel capped with a nitrogen outlet, and a rubber septum through which is inserted a temperature probe. The flask is flushed with nitrogen for 5 min and is then charged with (S)-1-chloro-3-(Boc-amino)-4-phenyl-2-butanone (3) (7.5 g, 25 mmol), which is added through temporary removal of the addition funnel. Anhydrous ethanol (200 mL, Note 13) is added by syringe through a septum. The addition funnel is opened slightly to allow delivery via syringe of a 1.0 M solution of lithium tri-tert-butoxyaluminum hydride in tetrahydrofuran (52.5 mL, 52.5 mmol) (Note 14). The flask is cooled to −78 °C (internal temperature) in a dry ice-acetone bath. The contents of the addition funnel are then added dropwise over 20 minutes so that the internal temperature does not exceed −70 °C. The mixture is stirred at this temperature for 2 h. The cooling bath is removed and the internal temperature is allowed to rise to −20 °C. The reaction is quenched through the addition of 50 mL of Rochelle's salt solution in one portion (Note 15). The bath is removed and stirring is continued for 15 min. The reaction mixture is concentrated by rotary evaporation (25 °C, 20 mmHg) to provide two liquid phases of ~80 mL combined volume. The concentrated mixture is transferred to a 1-L separatory funnel. An additional 200 mL of Rochelle's salt solution is added and the mixture is extracted with one 500-mL portion and one 150-mL portion of ethyl acetate. The combined extracts are washed with two 200-mL portions of water and one 200-mL portion of brine. The solution is dried over sodium sulfate, filtered and concentrated by rotary evaporation (25 °C, 20 mmHg) to obtain the crude product as a light yellow solid. This material is dissolved with heating in a mixture of 30 mL of ethyl acetate and 10 mL of hexanes (70 °C). Upon cooling, a solid separates which is collected by filtration on a 100-mL fritted-glass Büchner funnel and dried overnight at 25 °C and 0.5 mmHg to afford 4 (5.44–5.56 g, 82–84% yield) (Note 16) as a white solid.
2. Notes
1.
Trimethylsulfoxonium iodide was purchased from the Aldrich Chemical Company, Inc. and used as received.
2.
Anhydrous tetrahydrofuran (<50 ppm water) was purchased from EM Science and used as received.
3.
Potassium
tert-butoxide (1.0 M in tetrahydrofuran) was purchased from Aldrich Chemical Company, Inc. and used as received.
4.
This heating step was essential when the starting ester was potentially epimerizable. If this step was omitted in the case of sulfur ylide
2, good chemical yields were obtained but the product was nearly racemic.
5.
Boc-L-Phenylalanine 4-nitrophenyl ester was purchased from Bachem Bioscience, Inc. and used as received.
6.
Ethyl acetate was purchased from EMD Chemicals, Inc. and was used as received.
7.
An analytical sample of compound
2 that was dried for 24 h at room temperature at 0.5 mmHg had the following properties: mp (DSC) 163 °C. [α]
D25 −31.8 (c = 1.00, MeOH).
1H NMR
pdf (CDCl
3, 400 MHz) δ: 1.41 (s, 9 H), 2.98 (m, 2 H), 3.25 (s, 3 H), 3.35 (s, 3 H), 4.28 (m, 2 H), 5.22 (d,
J = 8.8, 1 H), 7.15–7.31 (m, 5 H).
13C NMR
pdf (CDCl
3, 100 MHz) δ: 28.3, 39.7, 41.7, 42.0, 57.8, 69.4, 79.2, 126.4, 128.1, 129.5, 137.6, 155.1, 186.5. Anal. Calcd for C
17H
25NO
4S: C, 60.15; H, 7.42; N, 4.13; S, 9.44 Found: C, 60.21; H, 7.61; N, 4.06. TLC: dichloromethane/methanol (95:5) R
f = 0.35.
8.
Hydrogen chloride (4.0 M in dioxane) was purchased from Aldrich Chemical Company, Inc. and was used as received.
9.
Hexanes was purchased from EMD Chemicals, Inc. and was used as received.
10.
The multiple washes were required to completely remove an impurity (presumably co-product DMSO) that interfered with the crystallization of
3.
11.
The enantiomeric excess was determined to be >99% by supercritical fluid chromatography (Chiralcel OD-H, 250 × 4.6 mm, 5 μm particle size, 3% methanol in CO
2 mobile phase, 40 °C, 2 mL/min, 150 bar). Retention times for the (
R)- and (
S)-enantiomers were 6.2 and 6.8 min, respectively. Properties of
3 are as follows: mp (DSC) 103 °C. [α]
D25 = −43.3 (c = 1.00, MeOH).
1H NMR
pdf (400 MHz, CDCl
3) δ: 1.40 (s, 9 H), 2.94–3.12 (m, 2 H), 4.00 (d,
J = 16.2, 1 H), 4.18 (d,
J = 16.2, 1 H), 4.68 (m, 1 H), 5.10 (b, 1 H), 7.14–7.47 (m, 5 H).
13C NMR
pdf (CDCl
3, 100 MHz) δ: 28.3, 37.8, 47.5, 58.5, 80.6, 126.2, 129.0, 129.2, 135.7, 155.3, 201.5. Anal. Calcd for C
15H
20ClNO
3: C, 60.50; H, 6.77; N, 4.70. Found: C, 60.15; H, 6.81; N, 4.72. TLC: hexanes/ethyl acetate (2:1) R
f = 0.7.
12.
The checkers used overhead mechanical stirring.
13.
Anhydrous ethanol (<0.005% water) was purchased from Aldrich Chemical Company, Inc. and used as received.
14.
Lithium tri-
tert-butoxyaluminum hydride (1.0M in tetrahydrofuran) was purchased from Aldrich Chemical Company, Inc. and used as received.
15.
Rochelle's salt solution was prepared by dissolving potassium sodium tartrate (30 g) and potassium carbonate (3.0 g) in 270 mL of water.
16.
The enantiomeric excess of the epoxide
4 was >99% by chiral HPLC. Conditions: Chiralcel OD NP, 250 × 4.6 mm, 10 μm particle size, solvent (isocratic) 98% heptane, 1% methanol, 1% ethanol, flow rate 1.0 mL/min. Retention times were:
(R,R)-epoxide, 10.2 min;
(S,S)-epoxide (
4), 11.0 min. Compound
4 had the following properties: mp (DSC) 125 °C. [α]
D25 −8.3 (c = 1.00, MeOH).
1H NMR
pdf (400 MHz, CDCl
3) δ: 1.38 (s, 9 H), 2.75 (br m, 1 H), 2.80 (m, 1 H), 2.87 (m, 1 H), 2.95 (m, 2 H), 3.69 (m, 1 H), 4.46 (br s, 1 H), 7.2–7.4 (m, 5 H).
13C NMR
pdf (100 MHz, CDCl
3) δ: 28.2, 37.5, 46.7, 52.5, 53.2, 79.5, 126.6, 128.4, 129.4, 137.7, 155.2. Anal. Calcd for C
15H
21NO
3: C, 68.42; H, 8.04; N, 5.32. Found: C, 68.44; H, 8.19; N, 5.30.
Handling and Disposal of Hazardous Chemicals
The procedures in this article are intended for use only by persons with prior training in experimental organic chemistry. All hazardous materials should be handled using the standard procedures for work with chemicals described in references such as "Prudent Practices in the Laboratory" (The National Academies Press, Washington, D.C., 2011 www.nap.edu). All chemical waste should be disposed of in accordance with local regulations. For general guidelines for the management of chemical waste, see Chapter 8 of Prudent Practices.
These procedures must be conducted at one's own risk. Organic Syntheses, Inc., its Editors, and its Board of Directors do not warrant or guarantee the safety of individuals using these procedures and hereby disclaim any liability for any injuries or damages claimed to have resulted from or related in any way to the procedures herein.
3. Discussion
N-Protected α-amino epoxides are broadly useful synthetic intermediates and in particular are ideal precursors for the hydroxyethylamine class of protease inhibitors. These compounds are frequently synthesized by one-carbon homologation of α-amino acids using diazomethane.
2 There are significant hazards associated with the use of diazomethane since this gaseous reagent is highly toxic, shock-sensitive, and explosive.
3 The procedure reported here, which is based on an earlier synthesis of α-fluorinated chloroketones,
4 was developed to provide an alternative to the use of diazomethane.
5
Efficient reaction of Corey's reagent, dimethylsulfoxonium methylide,
6 with esters requires an electron-withdrawing group adjacent to the carbonyl, provided in the case of
1 by the α-BOC-amino substituent. Several examples of chloroketone synthesis via this method are shown in the Table. Esters of simple aliphatic acids react only sluggishly. In the presence of a suitable activating group, methyl esters as well as
p-nitrophenyl esters will react. However, for substrates that are subject to epimerization such as
1, the more reactive aryl ester is needed to preclude partial racemization.
Our earlier procedure
5 for reduction of chloroketone
3 utilized sodium borohydride as the reductant. With that reducing agent, the
anti/syn selectivity was only 4:1, necessitating the isolation and crystallization of the intermediate β-chlorohydrin prior to epoxide ring-closure. The current procedure takes advantage of Hoffman's report
7 that high
anti-selectivity could be achieved with related substrates using lithium tri-
tert-butoxyaluminum hydride in ethanol. This protocol improved selectivity for reduction of
3 to 92:8 (
anti/syn). The intermediate β-chlorohydrins underwent epoxide ring-closure during work-up with the basic Rochelle salt solution. The desired
anti isomer
4 was isolated from the mixed diastereomers via a simple crystallization. The Rochelle's salt solution also served to sequester the aluminum-containing co-products, simplifying the extractive workup.
As an alternative to this “telescoped” procedure, it is also possible to isolate the β-chlorohydrin intermediate. This is accomplished by replacing the Rochelle salt solution with water during the extractive work-up.
5 The β-chlorohydrin is recovered as a mixture of diastereomers as shown below. The pure
anti-diastereomer is a crystalline solid (mp 122 °C) and can be isolated by crystallization from hot ethyl acetate.
5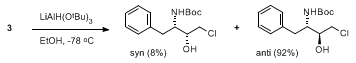
The best method for this reduction step varies depending on the nature of the α-chloroketone. When the chloroketone is prepared from an α-amino acid with a sterically large R group (e.g., valine), simple borohydride reduction provides high diastereoselectivity.
8 When the R group is small (e.g., alanine), hydrogenation using a “matched” ruthenium-BINAP catalyst is advantageous.
5 For asymmetric reduction of achiral chloroketones, the Noyori catalytic transfer hydrogenation procedure is recommended.
9 It should also be noted that, depending on which enantiomer of the Noyori catalyst is employed, this procedure can also be used to convert
3 into
either the
syn or the
anti β-chlorohydrin, in each case with 90:10 diastereoselectivity.
10 In cases where the final epoxide is not crystalline, it will generally be desirable to isolate and crystallize the intermediate β-chlorohydrin.
Appendix
Chemical Abstracts Nomenclature (Collective Index Number);
(Registry Number)
Trimethylsulfoxonium iodide; (1774-47-6)
Boc-L-phenylalanine 4-nitrophenyl ester:
L-Phenylalanine,
N-[(1,1-dimethylethoxy)carbonyl]-,
4-nitrophenyl ester; (7535-56-0)
Dimethylsulfoxonium (S)-2-oxo-3-(Boc-amino)-4-phenylbutylide:
Sulfoxonium, dimethyl-,
(3S)-3-[[(1,1-dimethylethoxy)carbonyl]amino]-2-oxo-4-phenylbutylide: (400611-25-8)
(S)-1-Chloro-3-(Boc-amino)-4-phenyl-2-butanone:
Carbamic acid,
[(1S)-3-chloro-2-oxo-1-(phenylmethyl)propyl]-,
1,1-dimethylethyl ester; (102123-74-0)
Lithium tri-tert-butoxyaluminum hydride; (17476-04-9)
(2S,3S)-1,2-Epoxy-3-(Boc-amino)-4-phenylbutane:
Carbamic acid,
[(1S)-1-(2S)-oxiranyl-2-phenylethyl]-,
1,1-dimethylethyl ester; (98737-29-2)
 |
William A. Nugent received his B. S. in Chemistry from Purdue University in 1969. After three years of teaching high school chemistry, he returned to Indiana University to earn his Ph. D. under the direction of Prof. Jay K. Kochi. In 1976 he joined the Central Research Department of the DuPont Company, later moving to the DuPont Pharmaceutical Company. Bristol-Myers Squibb subsequently acquired the DuPont Pharmaceutical Company and Bill currently holds the position of Senior Research Fellow at BMS. His principal research interest is the application of homogeneous catalysis in pharmaceutical manufacture. |
 |
Dengjin Wang was born in China in 1958. He received his B.S. degree from Nanjing University in 1982 and M.S. degree in 1987. In 1997, he joined DuPont-Merck Pharmaceuticals in the Department of Process Research and Development, where he worked on the development of anti-HIV agents. In 2001, he joined Bristol-Myers Squibb under the supervision of Dr. William Nugent. His research interests include catalysis and asymmetric reactions for C-C and C-N bond formation, as well as the development of practical processes for the large-scale production of active pharmaceutical ingredients and bioactive building blocks. |
 |
Jason Kowal earned an A.A.S degree in Chemical Technology in 1994 from Erie County Community College North Buffalo, New York followed by a B.S degree in Medicinal Chemistry in 1997 from State University of New York at Buffalo where his research began under Michael R. Detty, and finished under David G. Hangauer. Jason has been employed at Merck & Co., Inc. Process Research since 1997 and is currently a Research Chemist responsible for the development and implementation of new and efficient syntheses of novel active pharmaceutical ingredients from milligram to gram laboratory scale, to multi kilo bulk preparatory lab deliveries for clinical study, to pilot plant production. |
Copyright © 1921-, Organic Syntheses, Inc. All Rights Reserved