Org. Synth. 2010, 87, 95
DOI: 10.15227/orgsyn.087.0095
SYNTHESIS OF SUBSTITUTED INDAZOLES VIA [3+2] CYCLOADDITION OF BENZYNE AND DIAZO COMPOUNDS
[1H-indazole-3-carboxylic acid, ethyl ester]
Submitted by Feng Shi and Richard C. Larock
1.
Checked by Alistair Boyer and Mark Lautens.
1. Procedure
CAUTION! Due to the potential explosive nature of diazo compounds, the reaction should be conducted with proper precautions. A safety shield in a closed fume hood is recommended.
1H-Indazole-3-carboxylic acid, ethyl ester. A 1-L, flame-dried (Note 1), round-bottomed, three-necked flask (the central neck is sealed with a rubber septum, the other necks are sealed with a ground glass stopper and a tap adaptor attached to a nitrogen line) equipped with a large, egg-shaped stirring bar is charged with 2-(trimethylsilyl)phenyl trifluoromethanesulfonate (9.84 g, 32.0 mmol, 1.0 equiv) (Note 2) and ethyl diazoacetate (5.51 mL, 48.3 mmol, 1.5 equiv) (Note 3). Tetrahydrofuran (THF, Note 2) (360 mL) is introduced by cannula into the flask and the reaction mixture is cooled to -78 ºC (bath temperature) in an acetone/dry ice bath. Upon vigorous stirring, a TBAF solution (57.6 mL of 1 m THF solution, 57.6 mmol, 1.8 equiv) (Note 4) is added dropwise via syringe over ca. 40 min (Note 5). After complete addition, the reaction mixture is stirred at -78 ºC in the acetone/dry ice bath for 1.5 h. The flask is then transferred to a cold-acetone bath (ca. -65 °C, bath temperature) and allowed to warm to room temperature overnight, over which time the reaction mixture becomes orange. After a further 12 h stirring at room temperature, the reaction is judged complete (Note 6). The reaction mixture is concentrated at room temperature by rotary evaporation (35 °C, 40 mmHg) to ca. 100 mL and poured into a 1-L separatory funnel containing EtOAc (150 mL) and saturated aq. NaHCO3 (200 mL). The flask is rinsed with EtOAc (20 mL), which is added to the separatory funnel. The layers are separated, and the aqueous layer is extracted (2 × 50 mL) with EtOAc. The combined EtOAc extracts are dried over MgSO4 (ca. 20 g for 30 min), filtered through a fritted glass funnel, rinsing twice with EtOAc (20 mL), and evaporated to give an orange oil (ca. 27.6 g). The residue is purified by chromatography on SiO2 (Note 7) to afford 4.98 g (26.2 mmol, 82%) of 1H-indazole-3-carboxylic acid, ethyl ester as an off-white solid (Notes 8, 9).
2. Notes
1.
Although benzyne is highly reactive, the reaction summarized in this procedure does not exhibit air sensitivity. Thus, all weighing and transferring procedures can be carried out in air without a problem. The reaction does exhibit some, although not significant, moisture sensitivity, so drying the glassware and preventing the reaction mixture from absorbing moisture from outside air are desirable. It should be noted, however, that commercial TBAF solution contains about 5 wt% of water.
2.
The following chemicals were obtained from Aldrich and were used as received: ethyl diazoacetate; 2-(trimethylsilyl)phenyl trifluoromethanesulfonate (97%); and tetrabutylammonium fluoride (1.0 m solution in THF). The THF solvent (reagent grade, ~0.05% H
2O, obtained from Caledon Laboratories Ltd.) was dried over KOH then distilled from Na/benzophenone ketyl. All solvents used in the work-up procedures were standard reagent grade. Aluminium-backed TLC plates (silica gel, 60 F
254) were purchased from EMD Chemicals and 60 Å, 40-63 μm silica gel was purchased from Silicycle (submitters used 0.25 mm thick glass-backed TLC plates and 60 Å, 230-400 mesh silica gel purchased from Sorbent Technology).
3.
Commercial ethyl diazoacetate contains a small quantity of dichloromethane. The presence of dichloromethane does not hamper the reaction of ethyl diazoacetate with benzyne, but the added weight of dichloromethane should be taken into account. A
1H NMR spectrum should be taken prior to the use of this compound to determine the molar ratio of ethyl diazoacetate to dichloromethane. For both checkers and submitters, this ratio was 100:13. Thus, per 1.00 g of commercial ethyl diazoacetate (corresponding to 0.91 mL, based on d = 1.10 g/mL), 0.91 g of pure ethyl diazoacetate was present.
4.
The quality of commercial TBAF solutions can be variable. The checkers required 1.8 equiv TBAF whereas the submitters needed only 1.2 equiv. However, if incomplete conversion is observed, the reaction mixture can be re-cooled to -78 ºC and the protocol repeated using supplementary TBAF without any decrease of overall yield.
5.
The needle should be positioned a sufficient distance above the reaction mixture to prevent freezing of the solution in the needle and such that the TBAF solution is added directly to the reaction mixture to prevent freezing of the solution on the cold flask wall.
6.
A small aliquot (ca. 0.1 mL) of the reaction mixture was removed and concentrated
in vacuo. Direct
1H NMR (CDCl
3) analysis of this sample revealed the reaction mixture composition. Alternatively, a TLC analysis can be performed using 4:1 hexanes/EtOAc as the eluent. Unreacted starting material appears at R
f 0.65, a by-product (
1H-indazole-3-carboxylic acid, 1-phenyl, ethyl ester, blue visualized by short-wave UV) appears at
Rf 0.59, and the desired product appears at R
f 0.10.
7.
Column chromatography is performed on
400 mL of SiO2 (~160 g) packed into a 5.5 cm × 18 cm column with 3:1 hexanes/EtOAc. The crude material is loaded directly onto the packed SiO
2, washing with 50 mL of the eluent. Elute with 1.4 L of 3:1 hexanes/EtOAc, then change to 0.8 L of 1:3 hexanes/EtOAc, and collect 70 mL fractions throughout. The high running spots eluting together (1.78 g, fractions 7-16) contain a mixture of unreacted
ethyl diazoacetate (1.55 g, 13.6 mmol), unreacted
2-(trimethylsilyl)phenyl trifluoromethanesulfonate (0.12 g, 0.40 mmol, 1%) and the by-product:
1H-indazole-3-carboxylic acid, 1-phenyl, ethyl ester (0.11 g, 0.41 mmol, 3%). The low-running spot is the desired product (fractions 17-31), these fractions are concentrated by rotary evaporation (35 °C, 100 mmHg).
8.
This material is analytically pure, however, the color can be removed by crystallization.
Toluene (20 mL) is added to the colored product (4.98 g) in a single-necked, round-bottomed flask. The flask is fitted with a reflux condenser and the mixture is heated under reflux until the solid has dissolved (ca. 5 min). The flask is allowed to cool to ambient temperature over 1 h. The flask is transferred to a freezer (temperature -19 °C) for 10 h and the product is collected by filtration, washing with ice-cold
toluene (2 × 5 mL) and dried
in vacuo to give a white crystalline solid (4.71 g).
1H NMR analysis of the concentrated mother liquor reveals only the product and trace impurities.
9.
Data for product: R
f 0.30 (1.5:1 hexanes/EtOAc); mp 133-134 ºC (lit.
9 130 °C); IR (neat) 3292 (s), 1713 (s), 1479 (m), 1421 (m), 1273 (s), 1231 (s), 1140 (m) cm
-1;
1H NMR
pdf (400 MHz, CDCl
3) δ: 1.48 (3 H, t,
J = 7.1 Hz, CH
3), 4.56 (2 H, q,
J = 7.1 Hz, CH
2), 7.34 (1 H, t,
J = 7.5 Hz, Ar), 7.47 (1 H, t,
J = 7.6 Hz, Ar), 7.75 (1 H, d,
J = 8.5 Hz, Ar), 8.23 (1 H, d,
J = 8.2 Hz, Ar), 12.14 (1 H, br s, NH);
13C NMR
pdf (100 MHz, CDCl
3) δ: 14.4 (CH
3), 61.1 (CH
2), 111.1 (Ar-H), 121.8 (Ar-H), 122.4 (Ar), 123.2 (Ar-H), 127.3 (Ar), 127.3 (Ar-H), 141.4 (Ar), 163.0 (C=O); LRMS (EI) 88 (21), 90 (26), 118 (100), 145 (100), 162 (22), 190 (M
+, 66); HRMS (ESI
+)
m/z calcd. for C
10H
11N
2O
2 ([M+H]
+) 191.0815; found 191.0819; Anal. calcd. for C
10H
10N
2O
2: C, 63.15; H, 5.30; N, 14.73; found: C, 62.92; H, 5.11; N, 14.76.
Handling and Disposal of Hazardous Chemicals
The procedures in this article are intended for use only by persons with prior training in experimental organic chemistry. All hazardous materials should be handled using the standard procedures for work with chemicals described in references such as "Prudent Practices in the Laboratory" (The National Academies Press, Washington, D.C., 2011 www.nap.edu). All chemical waste should be disposed of in accordance with local regulations. For general guidelines for the management of chemical waste, see Chapter 8 of Prudent Practices.
These procedures must be conducted at one's own risk. Organic Syntheses, Inc., its Editors, and its Board of Directors do not warrant or guarantee the safety of individuals using these procedures and hereby disclaim any liability for any injuries or damages claimed to have resulted from or related in any way to the procedures herein.
3. Discussion
Benzyne is a highly reactive intermediate that has attracted wide attention from synthetic organic chemists recently.
2 Due to the high reactivity of benzyne, it is generated
in situ from various precursors. Two widely used precursors of benzyne are 2-(trimethylsilyl)phenyl trifluoromethanesulfonate
3 and (phenyl)[2-(trimethylsilyl)phenyl]iodonium trifluoromethanesulfonate.
4 The first compound and a few derivatives are now commercially available. These benzyne precursors undergo fluoride-promoted
ortho-elimination to generate benzyne under mild reaction conditions with a range of organic functional groups tolerated.
Benzyne has been shown to react with 1,3-dipoles under mild conditions to afford various important heterocycles.
5-7 Among these, reactions with diazo compounds afford substituted indazoles efficiently. It has been shown that an excess of a monosubstituted diazo compound reacts with benzyne as the limiting reagent to afford a 1
H-indazole after a hydrogen shift, as seen in the title reaction. Reaction with an excess of benzyne furnishes the product of
N-arylation. Thus, the title reaction can be performed on a 2.2:1 stoichiometry using cesium fluoride as the fluoride source in anhydrous acetonitrile at room temperature for 1 day to afford
N-arylated product (1
H-indazole-3-carboxylic acid, 1-phenyl, ethyl ester) in high yield (Eq 1).
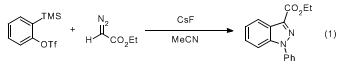
On the other hand, disubstituted diazo compounds can react with benzyne to afford 1
H-indazoles and/or 3
H-indazoles, depending upon the nature of the diazo compound and benzyne precursor. The product can be hard to predict.
5b Specifically, diazo compounds containing an acyl group may undergo acyl migration to afford 1
H-indazoles, as shown in Eq 2.
It has been established that acyclic ketone groups migrate preferentially and acyclic ester groups only migrate occasionally. Amide groups and aryl groups have not been observed to migrate. Cyclic diazo compounds containing acyl groups do not seem to react with benzynes under these conditions (Table 1).
It should be noted that these reactions perform much better on a small scale, often giving near quantitative yields. Once scaled up, these reactions tend to become dirty and give lower yields. In the reaction of Eq 2 mentioned above, we have observed an 85-90% yield on a 0.3-4.0 mmol scale; however, on a 20 mmol scale, this reaction gives incomplete conversion and only a 70% yield. A significant by-product is
III, the product of Eq 1, apparently formed by
N-deacylation, followed by
N-arylation. We reason that the acyl migration step may be responsible for this observation. The acyl migration has been suggested to proceed by a dissociation-ion pair intermediate-recombination process (Scheme 1).
8 In such a process, the [3+2] cycloaddition reaction should directly form intermediate
I, which dissociates to ion pair
IIa/b, which can recombine to form the desired product. The formation of the by-product
III is likely due to the reaction of the ion pair
IIb with free benzyne. However, the
N-deacylation of
IV by either trace amounts of moisture or the equilibrium between
IV and
IIb, followed by reaction with benzyne, cannot be ruled out. A control experiment has confirmed that isolated
IV reacts with benzyne under the same conditions to form a significant amount of
III. A similar observation has been made during the reaction of triethyl diazophosphonoacetate with benzyne, in which case the phosphonate group is lost and the product formed is again
III.
5bTable 1. Reaction of disubstituted diazo compounds with benzynes
Scheme 1. Proposed mechanism for acyl migration and by-product formation.
Appendix
Chemical Abstracts Nomenclature (Collective Index Number);
(Registry Number)
Ethyl diazoacetate; Acetic acid, diazo-, ethyl ester; (623-73-4)
2-(Trimethylsilyl)phenyl trifluoromethanesulfonate; Methanesulfonic acid, trifluoro-, 2-(trimethylsilyl)phenyl ester;; (88284-48-4)
Tetrabutylammonium fluoride; Butanaminium, N,N,N-tributyl- fluoride; (429-41-4)
 |
Richard C. Larock received his B.Sc. at the University of California, Davis in 1967. He then joined the group of Prof. Herbert C. Brown at Purdue University, where he received his Ph.D. in 1972. He worked as an NSF Postdoctoral Fellow at Harvard University in Prof. E. J. Corey's group and joined the Iowa State University faculty in 1972. His current research interests cover aryne chemistry, electrophilic cyclization, palladium catalysis, and polymer chemistry based on biorenewable resources. |
 |
Feng Shi was born in 1978 in China. He obtained his B.Sc. at Beijing University, China in 2001. Afterwards he moved to Michigan State University under the supervision of Prof. Robert E. Maleczka, Jr., where he received his Ph.D. in 2007 on iridium-catalyzed C-H activation chemistry. He joined Prof. Richard C. Larock's group in 2007 working on benzyne annulation chemistry. He is currently an associate professor at Henan University, China. |
 |
Alistair Boyer was born in 1982 in Warrington, UK. He obtained his M. Sci. at the University of Cambridge in 2004, completing his master project with Prof. Andrew B. Holmes. He stayed at Cambridge to perform his Ph.D. studies under the supervision of Prof. Steven Ley working on the synthesis of azadirachtin. In 2009, he moved to Toronto, Canada to become a post-doctoral research associate in the group of Prof. Mark Lautens, investigating novel rhodium-catalyzed reactions. |
Copyright © 1921-, Organic Syntheses, Inc. All Rights Reserved