Org. Synth. 2010, 87, 231
DOI: 10.15227/orgsyn.087.0231
SYNTHESIS OF YNAMIDES BY COPPER-MEDIATED COUPLING OF 1,1-DIBROMO-1-ALKENES WITH NITROGEN NUCLEOPHILES. PREPARATION OF 4-METHYL-N-(2-PHENYLETHYNYL)-N-(PHENYLMETHYL)BENZENESULFONAMIDE
Submitted by Alexis Coste, François Couty, and Gwilherm Evano
1.
Checked by Thomas P. Willumstad and Rick L. Danheiser.
1. Procedure
A. 4-Methyl-N-(phenylmethyl)benzenesulfonamide (2). An oven dried, 1-L, three-necked, round-bottomed flask equipped with a magnetic stirring bar (5 cm Teflon coated ovoid-shaped), a 250-mL pressure-equalizing addition funnel fitted with an argon inlet, two rubber septa, and a thermocouple probe is sequentially charged with benzylamine (11.5 mL, 11.3 g, 105.3 mmol, 1.05 equiv), triethylamine (15.5 mL, 11.3 g, 111.2 mmol, 1.1 equiv), and 250 mL of dichloromethane (Notes 1 and 2). The solution is cooled to 2-4 °C (internal temperature) in an ice bath and a solution of p-toluenesulfonyl chloride (19.1 g, 100.2 mmol) (Note 3) in 100 mL of dichloromethane is added dropwise via the addition funnel over 45 min. During the addition, the internal temperature of the flask is maintained below 5 °C. The mixture is stirred for 30 min at 2-4 °C. The ice bath is then removed and the resulting solution is allowed to warm to 21 °C, stirred at this temperature for 13 h, and then quenched by addition of 1 M HCl (500
mL). The resulting mixture is transferred to a 1-L separatory funnel and the organic layer is separated and washed with 1 M HCl (500
mL), 1 M NaOH (500
mL), saturated aqueous NaCl solution (500
mL), dried over MgSO4 (15 g), filtered, and concentrated by rotary evaporation (25 °C, 25 mmHg). The resulting solid is dried under vacuum (0.2 mmHg, 6 h) to afford 25.6 g (98%) of 4-methyl-N-(phenylmethyl)benzenesulfonamide as a white solid (Note 4).
B. (2,2-Dibromoethenyl)benzene (4). An oven dried 1-L, three-necked, round-bottomed flask equipped with a magnetic stirring bar (5 cm Teflon coated ovoid-shaped), 250-mL pressure-equalizing addition funnel fitted with an argon inlet, two rubber septa, and a thermocouple probe is sequentially charged with benzaldehyde (10.0 mL, 10.4 g, 98.5 mmol), carbon tetrabromide (49.7 g, 149.9 mmol, 1.5 equiv), and dichloromethane (350
mL) (Notes 5 and 6). The solution is cooled to 2-4 °C (internal temperature) with an ice-bath, and a solution of triisopropyl phosphite (54.0 mL, 45.6 g, 218.9 mmol, 2.2 equiv) (Note 7) in dichloromethane (70
mL) is added dropwise via the addition funnel over 90 min. During the addition, the internal temperature of the flask is maintained below 5 °C. The reaction mixture is stirred for 30 min at 2-4 °C and then transferred to a 1-L, single-necked, round-bottomed flask and concentrated on a rotary evaporator (25 °C, 25 mmHg). The flask is equipped with a magnetic stirring bar (5 cm Teflon coated ovoid-shaped) and a water-cooled condenser. 12 M HCl solution (110
mL) and glacial acetic acid (110
mL) (Note 8) are successively added, the condenser is capped with a two-tap Schlenk adaptor connected to a bubbler and an argon/vacuum manifold, and the reaction mixture is heated at reflux in an oil bath (120 °C external temperature) for 14 h. The resulting mixture is then allowed to cool to room temperature, diluted with 100 mL of dichloromethane, and transferred to a 1-L separatory funnel. The organic layer is separated and successively washed with distilled water (3 × 500 mL) and then transferred to a 1-L, single-necked, round-bottomed flask equipped with a teflon coated ovoid shaped magnetic stirring bar. Distilled water (400
mL) is added, followed by portionwise addition of sodium carbonate (50 g, 0.47 mol) (Note 9). The contents of the flask are transferred to a 1-L separatory funnel, and the organic layer is separated and washed with saturated aqueous NaCl solution (200
mL), dried over MgSO4 (7 g), filtered, and concentrated on a rotary evaporator (25 °C, 25 mmHg). The resulting yellow liquid is transferred to a 50-mL single-necked, round-bottomed flask. The flask is equipped with a magnetic stirring bar and fitted with a Vigreux column (8 cm, 1-cm diameter). The yellow liquid is purified by fractional distillation (80-111 °C, 7 mmHg) (Note 10) to give 20.8-22.4 g (81-87%) of (2,2-dibromoethenyl)benzene as a pale yellow liquid (Note 11).
C. 4-Methyl-N-(2-phenylethynyl)-N-(phenylmethyl) benzene-sulfonamide (5). An oven-dried, 100-mL, single-necked, round-bottomed flask equipped with a magnetic stirring bar (Note 12) is successively charged with N-4-toluenesulfonylbenzylamine 2 (6.3 g, 24.1 mmol), cesium carbonate (31.0 g, 95.1 mmol, 4 equiv) (Note 13), copper(I) iodide (570 mg, 3.0 mmol, 0.12 equiv) (Note 14), and (2,2-dibromoethenyl)-benzene (9.5 g, 36.3 mmol, 1.5 equiv). The flask is equipped with a water-cooled condenser capped with an argon gas inlet, and evacuated (8 mmHg) and backfilled with argon three times. The gas inlet is replaced by a rubber septum with an argon inlet needle. N,N'-Dimethylethylenediamine (0.45 mL, 0.37 g, 4.2 mmol, 0.18 equiv) (Note 15) and 1,4-dioxane (70
mL) (Note 16) are successively added through the septum. The resulting green-brown slurry is heated in a pre-heated oil bath at 70 °C (bath temperature) and vigorously stirred for 24 h (Note 17). The reaction mixture is cooled to room temperature and filtered into a 250-mL, single-necked, round-bottomed flask through a 125-mL fritted filtration funnel containing a pad of silica gel (Note 18). The filtrate is washed three times with 50 mL of ethyl acetate and then concentrated on a rotary evaporator (25 °C, 25 mmHg). The resulting oily residue is dissolved in diethyl ether (50
mL) and pentane (50
mL), concentrated by rotary evaporation at 25 °C, 25 mmHg) and then at 0.2 mmHg for 1 h, and then placed at -20 °C in a freezer overnight where it slowly solidifies (Note 19). A stir bar (1.5 cm Teflon coated ovoid-shaped) is added to the flask, absolute ethanol (25
mL) is added, and the flask is equipped with a water-cooled condenser and heated with stirring in an oil bath at 90 °C (bath temperature) until all of the material dissolves. The flask is then allowed to cool to room temperature (Note 20) and then cooled at -20 °C in a freezer for 8 h. The product is isolated by vacuum filtration through a fritted glass funnel (3 cm diameter, porosity 3) and washed with pentane (2 × 20 mL), and then dried under vacuum (0.2 mmHg, 4 h) to afford 6.3 g (72%) of 4-methyl-N-(2-phenylethynyl)-N-(phenylmethyl)benzenesulfonamide as a pale beige solid (Notes 21 and 22).
2. Notes
1.
Benzylamine (99%) was purchased from Acros and used as received.
Triethylamine (99%) was obtained from Acros by the Submitters and from J. T. Baker by the Checkers and used as received.
2.
Dichloromethane (> 99.5%) was purchased from Carlo Erba by the Submitters and from Mallinckrodt by the Checkers and used as received.
3.
p-Toluenesulfonyl chloride (98 %) was purchased from Alfa Aesar and used as received.
4.
In a run carried out on half-scale, 12.4 g (95%) of 4-methyl-
N-(phenylmethyl)-benzenesulfonamide was obtained. 4-Methyl-
N-(phenylmethyl)benzenesulfonamide has the following physical properties: Mp 114-115 °C; IR (KBr) 3270, 1598, 1496, 1454, 1326, 1162, 1059, 876, 743 cm
-1;
1H NMR
pdf (500 MHz, CDCl
3) δ: 2.45 (s, 3 H), 4.12 (d,
J = 6.0 Hz, 2 H), 4.81 (t,
J = 6.0 Hz, 1 H), 7.18-7.22 (m, 2 H), 7.24-7.33 (m, 5 H), 7.77 (d,
J = 8.5 Hz, 2 H);
13C NMR
pdf (125 MHz, CDCl
3) δ: 21.7, 47.4, 127.4, 128.06, 128.08, 128.9, 129.9, 136.5, 137.0, 143.7; HRMS-DART-ESI
m/z calcd. for C
14H
14NO
2S ([M-H]
-) 260.0751, found 260.0748.
5.
Benzaldehyde (99%) was purchased from Aldrich and used as received.
6.
Carbon tetrabromide (98%) was purchased from Alfa Aesar. It was dissolved in
dichloromethane (60 g in 300 mL), dried over
MgSO4 (20 g), filtered, concentrated on a rotary evaporator (30 °C, 20 mmHg), and dried under high vacuum (0.5 mmHg, 1 h) prior to use.
7.
Triisopropyl phosphite (90%) was purchased from Alfa Aesar and used as received.
8.
Concentrated hydrochloric acid and glacial acetic acid were purchased from Alfa Aesar and Mallinckrodt respectively and used as received.
9.
Sodium carbonate was added in 10 portions.
10.
A forerun of approximately 3 mL is collected and discarded.
11.
(2,2-Dibromoethenyl)benzene has the following physical properties: IR (film) 3057, 304, 1703, 1595, 1493, 1445, 1270, 1075, 920, 863 cm
-1;
1H NMR
pdf (500 MHz, CDCl
3) δ: 7.34-7.43 (m, 3 H), 7.51 (s, 1 H), 7.54-7.58 (m, 2 H);
13C NMR
pdf (125 MHz, CDCl
3) δ: 89.8, 128.60, 128.63, 128.8, 135.5, 137.1; HRMS-DART-ESI
m/z calcd. for C
8H
6Br
2 (M
+) 259.8831, found 259.8827.
12.
The reaction mixture being heterogeneous, an ovoid-shaped magnetic stirring bar longer than 2 cm is preferred. The use of a smaller magnetic bar did result in a failure to stir the reaction mixture properly in some cases.
13.
Cesium carbonate (99.9%) was purchased from Alfa Aesar and used as received. Use of less than 4 equivalents of cesium carbonate led to reduced yields and to the formation of small amounts of unidentified by-products.
14.
Copper iodide (99.999%) was purchased from Aldrich and used as received. The Submitters report that the yield was not significantly different when 98% copper iodide (purchased from Aldrich and used without purification) was employed for the reaction.
15.
N,N'-Dimethylethylenediamine (99%) was purchased from Aldrich and used as received.
16.
1,4-Dioxane (>99%) was purchased from Mallinckrodt and purified by distillation over sodium/benzophenone ketyl and then degassed by three successive freeze-pump-thaw cycles.
17.
The progress of the reaction was monitored by thin layer chromatography. TLC analysis was conducted using silica gel plates (elution with 10% EtOAc/hexanes, visualization by UV and KMnO
4); R
f values: vinyl dibromide
4 R
f = 0.97, sulfonamide
2 R
f = 0.22, ynamide
5 R
f = 0.58.
18.
The pad is prepared by slurrying
silica gel (30 g) with ethyl acetate in the fritted funnel (125 mL, 8 cm diameter, porosity 3) and filtering off the excess ethyl acetate.
19.
The Submitters report that in some cases the crude residue crystallized after concentration from the diethyl ether/pentane solution. It can then be immediately recrystallized.
20.
The Checkers found that crystallization was best initiated at room temperature by scratching with a glass rod prior to cooling at -20 °C.
21.
In a run carried out on half-scale, 3.20 g (73%) of 4-methyl-
N-(2-phenylethynyl)-
N-(phenylmethyl)benzenesulfonamide was obtained. This compound has the following physical properties: Mp 82-84 °C; IR (KBr) 2232, 1597, 1495, 1361, 1169, 1090, 939, 772 cm
-1;
1H NMR
pdf (500 MHz, CDCl
3) δ: 2.46 (s, 3 H), 4.60 (s, 2 H), 7.25 (app. s, 5 H), 7.31-7.37 (m, 7 H), 7.81 (d,
J = 8.0 Hz, 2 H);
13C NMR
pdf (125 MHz, CDCl
3) δ: 21.9, 55.9, 71.5, 82.8, 123.0, 127.9, 127.9, 128.4, 128.5, 128.7, 129.1, 129.9, 131.3, 134.6, 134.8, 144.8. Anal. calcd. for C
22H
19NO
2S: C: 73.10; H: 5.30; N: 3.88; found: C: 73.29; H: 5.25; N: 3.93.
22.
In solid form, 4-methyl-
N-(2-phenylethynyl)-
N-(phenylmethyl) benzenesulfonamide has been found to be stable to storage for months even at room temperature.
Handling and Disposal of Hazardous Chemicals
The procedures in this article are intended for use only by persons with prior training in experimental organic chemistry. All hazardous materials should be handled using the standard procedures for work with chemicals described in references such as "Prudent Practices in the Laboratory" (The National Academies Press, Washington, D.C., 2011 www.nap.edu). All chemical waste should be disposed of in accordance with local regulations. For general guidelines for the management of chemical waste, see Chapter 8 of Prudent Practices.
These procedures must be conducted at one's own risk. Organic Syntheses, Inc., its Editors, and its Board of Directors do not warrant or guarantee the safety of individuals using these procedures and hereby disclaim any liability for any injuries or damages claimed to have resulted from or related in any way to the procedures herein.
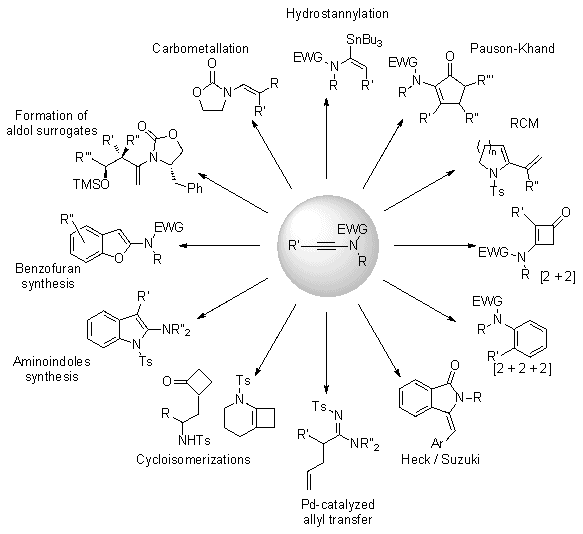
3. Discussion
Due to their superior stability compared to ynamines, ynamides are clearly emerging as key and versatile synthetic intermediates and have received considerable attention over the past decade.
2 They have been shown only quite recently to be excellent substrates for RCM,
3 palladium-mediated coupling reactions or rearrangements,
4 cycloisomerizations
5 and [2 + 2] cycloadditions,
6 radical transformations,
7 Pauson-Khand cyclizations,
8 diastereoselective sigmatropic rearrangements,
9 yne-carbonyl metathesis,
10 carbometalations,
11 and many other useful transformations. In addition, they have been used as efficient synthons for the preparation of a wide array of molecules such as, for example, keto-imides,
12 amino-indoles,
13 oxazolones,
14 indolines and carbazoles,
15 triazoles
16 or benzofurans (Figure 1).
17In addition to a number of traditionally employed approaches that often lack generality,
2 several strategies for the synthesis of ynamides have recently emerged. Although isomerization of propargyl amides or bromination/dehydrohalogenation of enamides represent facile entries to ynamides, they have been shown to be either substrate-specific or of too narrow scope. The first efficient procedure was based on the use of hypervalent alkynyliodonium salts which have been shown to cleanly react with metalated amides.
5b,8 This method is, however, limited to the preparation of ynamides substituted with either a trialkylsilyl or an aromatic group.
Figure 1. Utility of ynamides in organic synthesis
Recent advances in copper-catalyzed transformations provided the basis for new perspectives in ynamide synthesis. Inspired by the arylation of amides, Hsung and co-workers first developed a copper-catalyzed coupling between alkynyl bromides and amides, which provided an improved synthetic access to ynamides over existing protocols.
18 However, severe limitations remained, such as the use of high temperature and low substrate scope: although oxazolidinones were useful in the coupling, amides were mostly poor and sulfonamides were not suitable at all. In addressing this limitation, Danheiser and co-workers reported a useful solution using stoichiometric amounts of copper iodide along with KHMDS: this protocol allows reactions to proceed at room temperature with carbamates and sulfonamides but still requires the use of a strong base.
19 A general and quite mild procedure was finally published in 2004 by Hsung, who re-examined his coupling protocol by screening a variety of copper sources and ligands. The use of copper sulphate pentahydrate in combination with 1,10-phenanthroline proved to be especially successful, allowing the reaction to occur at temperatures ranging from 60 to 95 °C with potassium phosphate as base.
20Another option relies on the copper-catalyzed oxidative coupling between terminal alkynes and nitrogen nucleophiles recently reported by the Stahl group.
21 While good to excellent yields of various ynamides were obtained with this reaction, a major limitation is the use of five equivalents of the nucleophile, which can be prohibitive in some cases.
We have recently reported a new synthesis of ynamides based on the copper-mediated coupling between nitrogen nucleophiles and 1,1-dibromoalkenes.
22 This reaction has been shown to be general and provides a straightforward entry to ynamides that is complementary to previously reported synthetic routes. Ynamides are usually obtained in good yields and it is note worthy that this copper-catalyzed alkynylation of amides, sulfonamides, carbamates and oxazolidinones can be easily carried out on a gram or multigram scale (Figure 2). This method has the advantageous feature that it starts from readily available 1,1-dibromo-1-alkenes, which are attractive alkynylating agents. They can be prepared either using the classical Ramirez olefination
23 or the remarkably efficient Lautens modification,
24 which was used for the preparation of the vinyl-dibromide used in this procedure.
From a mechanistic point of view, we were able to demonstrate that the reaction proceeds through a regioselective monocoupling followed by dehydrobromination of the intermediate α-bromo-enamide which could be isolated in some cases by lowering the reaction temperature and cleanly transformed to the ynamide upon treatment with cesium carbonate.
25Figure 2. Scope of the copper-mediated coupling between nitrogen nucleophiles and 1,1-dibromoalkenes
Appendix
Chemical Abstracts Nomenclature (Collective Index Number);
(Registry Number)
4-Methyl-N-(phenylmethyl)benzenesulfonamide; (1576-37-0)
Benzylamine: Benzenemethanamine; (100-46-9)
p-Toluenesulfonyl chloride: Benzenesulfonyl chloride, 4-methyl-; (98-59-9)
Triethylamine: Ethanamine, N,N-diethyl-; (121-44-8)
(2,2-Dibromoethenyl)benzene; (7436-90-0)
Carbon tetrabromide: Methane, tetrabromo-; (558-13-4)
Triisopropyl phosphite: Phosphorous acid, tris(1-methylethyl) ester; (116-17-6)
Benzaldehyde; (100-52-7)
4-Methyl-N-(2-phenylethynyl)-N-(phenylmethyl)benzenesulfonamide: (609769-63-3)
Cesium carbonate; (534-17-8)
Copper(I) iodide; (1335-23-5)
N,N'-Dimethylethylenediamine: 1,2-Ethanediamine, N1,N2-dimethyl-; (110- 70-3)
 |
Gwilherm Evano was born in Paris in 1977 and studied chemistry at the Ecole Normale Supérieure in Paris and received his Ph.D from Université Pierre et Marie Curie in 2002 under the supervision of François Couty and Claude Agami. After postdoctoral study with James S. Panek at Boston University, he joined the CNRS as Chargé de Recherche at the University of Versailles in 2004. His research interests focus on asymmetric synthesis and reactivity of nitrogen heterocycles, copper-catalyzed transformations, and the total synthesis of natural and/or biologically relevant products. |
 |
Alexis Coste was born in 1982 and studied chemistry at the Ecole Supérieure de Chimie Organique et Minérale. Since 2007, he is engaged in Ph.D. research under the supervision of François Couty and Gwilherm Evano at the University of Versailles as a National Cancer Institute Fellow. His work focuses on the development of copper-catalyzed transformations with application in natural product synthesis and on the development of new proteasome inhibitors in a tumor targeting approach. |
 |
François Couty was born in Caen (France) in 1963. He studied chemistry at the University Pierre et Marie Curie in Paris and completed his Ph.D. with Claude Agami in 1991. The same year, he was appointed assistant professor in this University. He spent a year in Namur (Belgium) as a postdoctoral fellow with Prof. A. Krief. After having completed his Habilitation in 1999, he was promoted full Professor at the University of Versailles in 2001. His research interests are in the field of asymmetric synthesis, including the development of new synthetic methodologies for nitrogen heterocycles, the total synthesis of natural products and projects at the chemistry-biology interface. |
 |
Thomas Willumstad was born in 1985 in Pueblo, Colorado. In 2004, he received his B.A. degree in chemistry from the University of Colorado at Boulder. While at CU he worked on methods directed toward the synthesis of diazonamide A in the lab of Professor Tarek Sammakia. Tom is currently pursuing graduate studies at the Massachusetts Institute of Technology under the guidance of Professor Rick Danheiser. His research focuses on the synthesis of highly substituted quinolines via a tandem benzannulation/iodocylization strategy. |
Copyright © 1921-, Organic Syntheses, Inc. All Rights Reserved