Org. Synth. 2010, 87, 299
DOI: 10.15227/orgsyn.087.0299
PALLADIUM-CATALYZED ALKYL-ALKYL SUZUKI CROSS-COUPLINGS OF PRIMARY ALKYL BROMIDES AT ROOM TEMPERATURE: (13-CHLOROTRIDECYLOXY)TRIETHYLSILANE
[Silane, [(13-chlorotridecyl)oxy]triethyl-]
Submitted by Sha Lou and Gregory C. Fu
1.
Checked by Takuya Higo and Tohru Fukuyama.
1. Procedure
A. 1-Bromo-8-chlorooctane (1). An oven-dried, 200-mL, two-necked, round-bottomed flask equipped with an argon inlet and a magnetic stirbar (octagonal, molded pivot ring, 25 mm length and 6 mm diameter) is purged with argon for 5 min and then charged through the open neck with CH2Cl2 (50 mL via syringe) (Note 1), imidazole (2.19 g, 32.1 mmol, 1.10 equiv) (Note 2), and dichlorotriphenylphosphorane (10.4 g, 31.2 mmol, 1.07 equiv) (Note 3). The open neck is capped with a rubber septum, and the stirred solution is cooled in an ice bath for 5 min. A solution of 8-bromo-1-octanol (5.0 mL, 6.11 g, 29.2 mmol, 1.00 equiv) (Note 4) in CH2Cl2 (10
mL) (Note 1) is added via syringe over 5 min. The reaction mixture is allowed to warm to rt, and the resulting heterogeneous solution (a white precipitate formed) is stirred for 4 h. The progress of the reaction is followed by TLC analysis on SiO2 (10% EtOAc/hexanes as the eluent; visualization with a KMnO4 stain; the alcohol starting material has an Rf = 0.2, and the chloride product has an Rf = 0.9) (Note 5). After the alcohol is consumed, the reaction is diluted with pentane (200
mL), and the mixture is filtered through a pad of SiO2 (7 cm diameter × 6 cm height) in a sintered glass funnel. The SiO2 is washed with additional pentane (400
mL). The filtrate is concentrated by rotary evaporation (20 mmHg, 30 °C), which furnishes the desired product as a colorless oil (6.23-6.44 g, 94-97 % yield) (Note 6). The product is used in the next step without further purification.
B. Triethyl(pent-4-enyloxy)silane (2). An oven-dried, 200-mL, two-necked, round-bottomed flask equipped with an argon inlet and a magnetic stirbar (octagonal, molded pivot ring, 25 mm length and 6 mm diameter) is purged with argon for 5 min and then charged through the open neck with N,N-dimethylformamide (50 mL via syringe) (Note 7), 4-penten-1-ol (8.93 mL via syringe, 7.50 g, 87.1 mmol, 1.00 equiv) (Note 8), and imidazole (5.93 g, 87.1 mmol, 1.00 equiv) (Note 2). The open neck is capped with a rubber septum. The stirred solution is cooled in an ice bath for 5 min, and then chlorotriethylsilane (14.6 mL, 13.1 g, 87.1 mmol, 1.00 equiv) (Note 9) is added over 4 min via syringe. The reaction mixture is stirred at rt for 24 h. The progress of the reaction is followed by TLC analysis on SiO2 (20% EtOAc/hexanes as the eluent; visualization with a KMnO4 stain; the alcohol starting material has an Rf = 0.2, and the silyl ether product has an Rf = 0.7) (Note 5). After the alcohol has been consumed, the reaction mixture is poured into a mixture of pentane (300
mL) and water (60
mL) in a 500-mL separatory funnel. The organic layer is separated and washed with brine (3 × 50 mL). The organic solution is dried over MgSO4 (30 g) and then vacuum filtered through a Büchner funnel containing a bed of celite (1.0 cm height). The filtrate is concentrated by rotary evaporation (20 mmHg, 30 °C), and the residue is transferred to a 50-mL round-bottomed flask equipped with a magnetic stirbar (octagonal, molded pivot ring, 15 mm length and 7 mm diameter) and a short-path distillation head. The residue is distilled under vacuum (bp 77-79 °C at 8 mmHg), which provides the desired silyl ether 2 as a colorless oil (15.2-15.7 g, 87-90% yield) (Note 10).
C. (5-(9-Borabicyclo[3.3.1]nonan-9-yl)pentyloxy)triethylsilane (3). An oven-dried, 200-mL, round-bottomed flask equipped with an argon inlet and a magnetic stirbar (octagonal, molded pivot ring, 25 mm length and 6 mm diameter) is purged with argon for 10 min. The open neck is capped with a rubber septum, and then a solution of 9-borabicyclo[3.3.1]nonane (9-BBN; 0.50 M in THF; 72 mL, 36 mmol, 1.0 equiv) (Note 11) is added via syringe. Next, triethyl(pent-4-enyloxy)silane (2) (7.21 g, 36.0 mmol, 1.0 equiv) is added via syringe over 3 min to the solution of 9-BBN. The reaction mixture is stirred for 3 h, at which time all of the starting olefin is consumed as determined by TLC analysis (pentane as the eluent; visualization with a KMnO4 stain; the olefin starting material has an Rf = 0.2) (Note 5). This solution is used directly in the next step.
D. (13-Chlorotridecyloxy)triethylsilane (4). An oven-dried, 1000-mL, three-necked, round-bottomed flask equipped with a thermometer inlet, a thermometer, a magnetic stir bar (octagonal, molded pivot ring, 40 mm length and 10 mm diameter), and an argon inlet is purged with argon for 10 min. Palladium(II) acetate (270 mg, 1.20 mmol, 0.040 equiv) (Note 12), tricyclohexylphosphine (673 mg, 2.40 mmol, 0.080 equiv) (Note 13), and tripotassium phosphate, monohydrate (K3PO4•H2O; 8.28 g, 36.0 mmol, 1.2 equiv) (Note 14) are added through the open neck of the flask. Then, the open neck is capped with a rubber septum, and the solution of (5-(9-borabicyclo[3.3.1]nonan-9-yl)-pentyloxy)triethylsilane (3) prepared in Step C (36 mmol, 1.2 equiv) is added to the flask via syringe, followed by the addition of 1-bromo-8-chlorooctane (1) (6.83 g, 30.0 mmol, 1.0 equiv). The resulting dark-brown heterogeneous reaction mixture is stirred vigorously at rt for 24 h. The progress of the reaction is followed by TLC analysis on SiO2 (25% CH2Cl2/hexanes as the eluent; visualization with a KMnO4 stain; the alkyl bromide starting material has an Rf = 0.5, and the cross-coupling product has an Rf = 0.4) (Note 5). Next, the mixture is diluted with diethyl ether (200
mL) and filtered through a sintered glass funnel containing SiO2 (7.0 cm diameter × 5.0 cm height). The SiO2 is washed with additional diethyl ether
(200 mL), and the combined filtrate is concentrated by rotary evaporation (20 mmHg, 30 °C). The residue is purified by column chromatography on SiO2 (Note 15). The desired cross-coupling product 4 has Rf = 0.7 (TLC analysis on SiO2: 50% CH2Cl2/hexanes as eluent, visualization with KMnO4) (Note 5). The cross-coupling product is obtained as a pale-yellow oil (9.63-10.10 g, 92-96% yield) (Note 16).
2. Notes
1.
Dichloromethane (>99.5%) was purchased from Kanto Chemical Co., Inc. (water content <0.001%) and purified by Glass Contour solvent systems. The submitters purchased
dichloromethane (>99.8%) from J.T. Baker (water content <0.02%), which was purified by passage through activated alumina under argon.
2.
Imidazole (99%) was purchased from Alfa Aesar and used as received.
3.
Dichlorotriphenylphosphorane (95%) was purchased from Aldrich and used as received.
4.
8-Bromo-1-octanol (95%) was purchased from Alfa Aesar and used as received.
5.
Analytical thin-layer chromatography was performed using Merck silica gel 60 F254 plates (0.25 mm).
6.
Compound
1 has the following properties: IR (film): 2931, 2855, 1457, 1291, 1245, 913 cm
-1;
1H NMR
pdf (CDCl
3, 400 MHz) δ: 1.31-1.35 (m, 4 H), 1.40-1.47 (m, 4 H), 1.75-1.81 (m, 2 H), 1.82-1.89 (m, 2 H), 3.41 (t,
J = 7.0 Hz, 2 H), 3.54 (t,
J = 7.0 Hz, 2 H);
13C NMR
pdf (CDCl
3, 100 MHz) δ: 26.7, 28.0, 28.6, 28.7, 32.5, 32.7, 34.0, 45.1; Anal. Calcd. for C
8H
17BrCl: C, 42.22; H, 7.09; N, 0; found: C, 41.94; H, 6.85; N, 0. The spectral data are in agreement with the reported values.
2 Compound
1 can be purified via column chromatography on SiO
2, eluting with pentane (R
f = 0.6, pentane; visualization with KMnO
4).
7.
N,N-Dimethylformamide (>99.5) was purchased from Wako Pure Chemical Industries, Ltd. and used as received. The submitters purchased
N,
N-dimethylformamide (ACS reagent grade) from MP Biomedicals, LLC and used it as received.
8.
4-Penten-1-ol (98+%) was purchased from Alfa Aesar and used as received.
9.
Chlorotriethylsilane (98+%) was purchased from Alfa Aesar and used as received.
10.
Compound
2 has the following properties: IR (film): 2947, 2884, 2826, 1653, 1457, 1418, 1015, 743 cm
-1;
1H NMR
pdf (CDCl
3, 400 MHz) δ: 0.60 (q,
J = 8.0 Hz, 6 H), 0.96 (t,
J = 8.0 Hz, 9 H), 1.60-1.66 (m, 2 H), 2.11 (q,
J = 6.8 Hz, 2 H), 3.62 (t,
J = 6.4 Hz, 2 H), 4.94-5.05 (m, 2 H), 5.79-5.86 (m, 1 H);
13C NMR
pdf (CDCl
3, 100 MHz) δ: 4.4, 6.8, 30.1, 32.0, 62.3, 114.5, 138.5; LRMS (DART)
m/z calcd. for C
11H
25OSi ([M+H]
+) 201.2; found 201.2. The spectral data are in agreement with the reported values.
211.
The solution of 9-BBN (0.50 M in THF) was purchased from Aldrich and used as received.
12.
Pd(OAc)2 (99+%) was purchased from Strem and used as received.
13.
PCy3 (97%) was purchased from Strem and used as received.
14.
Tripotassium phosphate, monohydrate (≥94%) was purchased from Fluka and used as received. When anhydrous tripotassium phosphate is employed, essentially no cross-coupling is observed.
15.
Column chromatography was performed on Kanto Chemical Silica Gel 60N (wet packed in hexanes; 7 cm diameter × 13 cm height; 200 g), eluting with a gradient of CH
2Cl
2 in hexanes (500 mL of hexanes, 500 mL of 5% CH
2Cl
2/hexanes, 1.0 L of 10% CH
2Cl
2/hexanes, 1.0 L of 15% CH
2Cl
2/hexanes; 100-mL fractions). All of the fractions (8-26) containing the desired product were combined and concentrated by rotary evaporation (20 mmHg, 30 °C).
16.
Compound
4 has the following properties: IR (film): 2926, 2875, 2854, 1459, 1414, 1384, 1238, 1098, 1007, 913, 735 cm
-1;
1H NMR
pdf (CDCl
3, 400 MHz) δ: 0.60 (q,
J = 8.0 Hz, 6 H), 0.96 (t,
J = 8.0 Hz, 9 H), 1.26-1.28 (m, 16 H), 1.39-1.45 (m, 2 H), 1.50-1.56 (m, 2 H), 1.73-1.80 (m, 2 H), 3.53 (t,
J = 6.8 Hz, 2 H), 3.59 (t,
J = 6.4 Hz, 2 H);
13C NMR
pdf (CDCl
3, 100 MHz) δ: 4.4, 6.8, 25.8, 26.9, 28.9, 29.47, 29.54, 29.59, 29.60, 29.62, 32.6, 32.9, 45.2, 63.0; LRMS (DART)
m/z calcd. for C
19H
42ClOSi ([M+H]
+) 349.3, found 349.3; Anal. calcd. for C
11H
42OSi: C, 65.93; H, 12.07; N, 0; found: C, 65.99; H, 11.80; N, 0. The spectral data are in agreement with the reported values.
2
Handling and Disposal of Hazardous Chemicals
The procedures in this article are intended for use only by persons with prior training in experimental organic chemistry. All hazardous materials should be handled using the standard procedures for work with chemicals described in references such as "Prudent Practices in the Laboratory" (The National Academies Press, Washington, D.C., 2011 www.nap.edu). All chemical waste should be disposed of in accordance with local regulations. For general guidelines for the management of chemical waste, see Chapter 8 of Prudent Practices.
These procedures must be conducted at one's own risk. Organic Syntheses, Inc., its Editors, and its Board of Directors do not warrant or guarantee the safety of individuals using these procedures and hereby disclaim any liability for any injuries or damages claimed to have resulted from or related in any way to the procedures herein.
3. Discussion
The palladium-catalyzed coupling of organometallic compounds with aryl and vinyl halides is a now-classic method for carbon-carbon bond formation.
3 In contrast, until recently, the corresponding reactions of
alkyl halides were relatively uncommon.
3 Slow oxidative addition and facile ß-hydride elimination have been suggested as two of the possible culprits for this comparative lack of success (Scheme 1). With respect to the Suzuki reaction, prior to 2001 only one somewhat general method had been described for achieving cross-couplings of unactivated, β-hydrogen containing alkyl electrophiles, specifically, a Pd(PPh
3)
4-catalyzed process for coupling alkyl iodides.
5Scheme 1. A Generalized Mechanism for Palladium-Catalyzed Cross-Coupling of an Alkyl Electrophile.
We have determined that, through the appropriate choice of ligand, palladium-catalyzed Suzuki cross-couplings can be accomplished with an array of unactivated alkyl bromides, chlorides, and tosylates that bear β hydrogens.
2,6,7 Specifically, bulky electron-rich trialkylphosphines furnish particularly active catalysts. For example, in the case of alkyl bromides, Pd(OAc)
2/PCy
3 achieves the desired coupling under mild conditions (room temperature; Table 1). The process is compatible with a broad spectrum of functional groups, including amines, alkenes, esters, alkynes, ethers, and nitriles. Furthermore, an alkyl bromide can be cross-coupled selectively in the presence of an alkyl chloride (equation D). Not only alkylboranes (entries 1-6), but also vinylboranes (entry 7), serve as suitable coupling partners.
Table 1. Pd/PCy3-Catalyzed Suzuki Cross-Couplings of Unactivated Alkyl Bromides at Room Temperature.
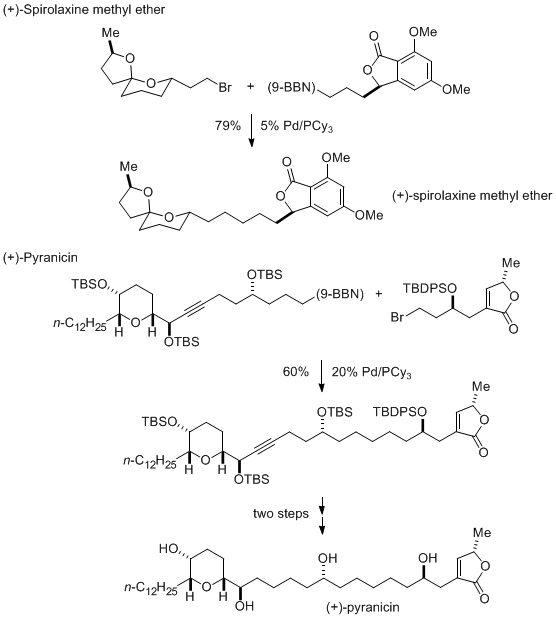
This method for Suzuki coupling of alkyl bromides has been employed by others, e.g., by Phillips to achieve late-stage fragment couplings in natural-product total synthesis (Scheme 2).
8Scheme 2. Applications in Total Synthesis of Pd/PCy3-Catalyzed Alkyl-Alkyl Suzuki Reactions: Fragment Couplings.
Appendix
Chemical Abstracts Nomenclature (Collective Index Number);
(Registry Number)
8-Bromo-1-octanol: 1-Octanol, 8-bromo-; (50816-19-8)
Dichlorotriphenylphosphorane: Phosphorane, dichlorotriphenyl-; (2526-64-9)
Imidazole: 1H-Imidazole; (288-32-4)
1-Bromo-8-chlorooctane: Octane, 1-bromo-8-chloro-; (28598-82-5)
4-Penten-1-ol; (821-09-0)
Chlorotriethylsilane: Silane, chlorotriethyl-; (994-30-9)
Triethyl(pent-4-enyloxy)silane: Silane, triethyl(4-penten-1-yloxy)-; (374755-00-7)
9-BBN: 9-Borabicyclo[3.3.1]nonane; (280-64-8)
(5-(9-Borabicyclo[3.3.1]nonan-9-yl)pentyloxy)triethylsilane: 9-Borabicyclo[3.3.1]nonane, 9-[5-[(triethylsilyl)oxy]pentyl]-; (157123-09-6)
Palladium(II) acetate: Acetic acid, palladium(2+) salt (2:1); (3375-31-3)
Tricyclohexylphosphine: Phosphine, tricyclohexyl-; (2622-14-2)
Tripotassium phosphate, monohydrate: Phosphoric acid, tripotassium salt, monohydrate (8CI,9CI); (27176-10-9)
(13-Chlorotridecyloxy)triethylsilane: Silane, [(13-chlorotridecyl)oxy]triethyl-; (374754-99- 1)
 |
Prof. Greg Fu was born in Galion, Ohio, in 1963. He received a B.S. degree in 1985 from MIT, where he worked in the laboratory of Prof. K. Barry Sharpless. After earning a Ph.D. from Harvard in 1991 under the guidance of Prof. David A. Evans, he spent two years as a postdoctoral fellow with Prof. Robert H. Grubbs at Caltech. In 1993, he returned to MIT, where he is currently the Firmenich Professor of Chemistry. Prof. Fu received the Springer Award in Organometallic Chemistry in 2001, the Corey Award of the American Chemical Society in 2004, and the Mukaiyama Award of the Society of Synthetic Organic Chemistry of Japan in 2006. He is a fellow of the Royal Society of Chemistry and of the American Academy of Arts and Sciences. Prof. Fu serves as an associate editor of the Journal of the American Chemical Society.His current research interests include metal-catalyzed coupling reactions, chiral-ligand design, and enantioselective nucleophilic catalysis. |
 |
Sha Lou was born in He Bei, China, in 1979. He received a B.S. in Chemistry from Beijing University of Chemical Technology in 2002, where he conducted undergraduate research on fullerene functionalizations with Professor Shenyi Yu. He obtained a Ph.D. degree in January 2008 from Boston University under the direction of Professor Scott E. Schaus. His graduate research focused on transition metal- and organic molecule-catalyzed asymmetric carbon-carbon bond-forming reactions and synthesis. In 2008, he joined the group of Professor Greg Fu at MIT as a postdoctoral fellow. His current research involves the development of transition metal-catalyzed enantioselective cross-coupling reactions. |
 |
Takuya Higo was born in Saga, Japan in 1986. He received his B.S. in 2009 from the University of Tokyo. In the same year, he began his graduate studies at the Graduate School of Pharmaceutical Sciences, the University of Tokyo, under the guidance of Professor Tohru Fukuyama. His research interests are in the area of the total synthesis of natural products. |
Copyright © 1921-, Organic Syntheses, Inc. All Rights Reserved